Rapid Filter or Resin Change Strategies for Biomanufacturing

Pandemic-related supply chain shortages have placed constraints on the supply of essential filters and chromatography resins. An agile regulatory pathway to implement alternative filters and resins into manufacturing is necessary to ensure the continued supply of approved biologics. To allow this in the US and potentially globally, the regulatory strategy proposed in this article is to provide an appropriate characterization package to demonstrate that the alternative filter or resin has a low risk to impact product quality in a prior approval supplement (PAS), and later provide at-scale data as part of an annual report or submission at the time of distribution.
Both the development of COVID-19 vaccines and monoclonal antibody therapies and supply chain shortages related to the pandemic have affected sup-ply of the essential filters and chromatography resins used in the manufacture of biological products. Section 101 of the Defense Production Act of 1950 was extended by Executive Order 13911 in 2020 to Respond to the Spread of COVID-19. This has allowed for regulation of the allocation of necessary supplies to the production of COVID-related therapies through rated orders.1 The unpredictable timing of rated orders has placed constraints on filter vendors because they are unable to confirm delivery of orders needed to support manufacture of clinical and commercial biologic therapies not targeting COVID-19.2, 3
Since the start of the pandemic, companies have employed multiple risk mitigation measures to conserve supply of filters and chromatography resins. However, given the continued uncertainty in supply, use of alternative filters or resins may be necessary to ensure continued patient supply of approved biologics. For critical filters and resins in manufacturing of biological therapeutics, each health authority typically requires approval before final product distribution using alternatives.
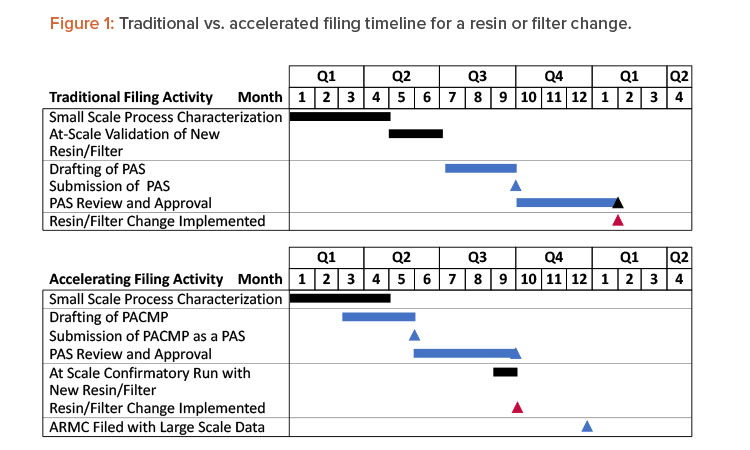
This article presents a proposed regulatory strategy to help alleviate supply risks for approved therapeutics. The strategy is to provide an appropriate characterization package that demonstrates the alternative filter or resin will not impact product quality, submit it in a PAS, and, if needed, submit a request for an expedited review. The initial PAS would also include a commitment to provide any at-scale product quality and/or validation data in the US Annual Report of Minor Changes (ARMC) or potentially a submission at the time of distribution (changes being effected [CBE-0]). Acceptance criteria for confirmatory at-scale data would be proposed in the PAS as part of the postapproval change management protocol (PACMP). This strategy saves approximately 4 to 6 months (depending on the time required for PAS approval) before initiation of the resin and filter change, as shown in Figure 1. With the endorsement of International Council for Harmonisation (ICH) Q12 in November 2019,4 and its ongoing implementation in ICH member countries, applying this approach has the potential for use as a global regulatory strategy.
The proposed characterization and validation plan is adequate to support approval of an alternate:
- Sterilizing-grade filter option for aseptic drug product manufacturing
- Viral filter (and potentially viral pre-filter) for drug substance manufacturing
- Ultrafiltration and diafiltration (UF/DF) membrane for drug substance manufacturing
- Protein A affinity chromatography resin for drug substance manufacturing
The proposed submission category of annual reportable for the protein A chromatography resins, filters, and other single-use raw materials is presented next. In all cases, these changes are enabled by strong product and process knowledge and mature quality systems.3, 5
Though not the focus here, when scientifically justified, the characterization package for a given resin or filter may be used to support more than one product.3, 5 Similarly, prior knowledge from large- or small-scale characterization or validation studies with multiple earlier products for a particular resin or filter could be used to support a current product. Details on the proposed characterization and at-scale studies to support alternative filters and resins are provided later in this article.
Background
Companies have experienced significant procurement delays for filters and resins because of the COVID-19 pandemic. Because of rated orders and resulting prioritization, some vendors have stopped or delayed confirmation of orders, delayed previously confirmed orders, and significantly delayed deliveries. Purchase orders that were placed up to a year prior, including previously confirmed orders, are not being delivered with little to no advance notice.
Companies have proactively managed these risks to avoid drug shortages. In late 2020, many companies evaluated mitigation options across commercial and clinical portfolios to slow consumption of critical at-risk raw materials in response to early signals from suppliers on the potential impact of rated orders on their supply chains. Since the start of the pandemic, the Center for Drug Evaluation and Research (CDER) has received about 150 inquiries related to strategies for chemistry, manufacturing, and control (CMC) changes, including changes to filter suppliers, filter control strategies, and filter reuse validation.5
The FDA has indicated it may “consider available information and approaches to mitigate the risk to product quality associated with the change to support a reporting category for certain supplements that is lower than what otherwise would be most suitable.”6In addition, the FDA is using multiple tools to facilitate implementation of manufacturing changes, such as risk-based reduction in supplement reporting categories and flexible assessment practices.3, 6 With these tools, the rationale, supporting information, and risk analysis should be provided.
Within the drug substance and drug product manufacturing processes, filters are used for a variety of purposes, including non-product contact (e.g., media, buffer, and air/vent filters); product contact for process and product impurity; and bioburden and particle removal. Similarly, chromatography resins play an important role as the primary means of impurity removal and potentially viral clearance within the drug substance manufacturing process.
Given the criticality of chromatography resins and some product contact filters in meeting product quality acceptance criteria and specifications, significant process characterization is performed before a product contact filter or resin is selected. Demonstration of performance is included in viral clearance and process validation, which are submitted as part of product licensure. These characterization and qualification activities can require more than 6 months to complete.
Currently, companies are employing multiple strategies to proactively mitigate against the risks presented by resin or filter supply delays or disruptions, for example:
- Communicating critical resin and filter demand requirements to suppliers
- Developing an approach to extend expiry and the number of reuses for reusable resins and filters, such as tangential flow filtration (TFF)
- Pursuing alternate sourcing and substituting at-risk filters/resins with alternates
- Adjusting manufacturing schedules to accommodate late deliveries of resins and/or product contact filters
Given the uncertainty of how long and to what extent resin and product contact filter supply delays may continue, companies intend to implement alternative resins and filters to mitigate risk to product supply and ensure continued supply to patients in need.
Discussion
Several different protein A affinity resins are commercially available that leverage the highly selective binding of immobilized protein A ligands to the Fc region of monoclonal antibodies and other Fc-containing molecules (e.g., fusion proteins). The majority of the studies performed to qualify an alternate protein A resin can be completed using a suitable scale-down model before introducing it into full-scale production (e.g., process characterization; process linkage, cleaning, and reuse). Essential performance characteristics can also be evaluated by those with prior knowledge (including platform knowledge) using additional small-scale studies before full-scale production.
Filters and other single-use components are essential to production of biological therapeutics. Although their importance is paramount to ensuring supply of quality product, their function and critical requirements are relatively well-understood. Alternate suppliers of equivalent filters/components with comparable critical material attributes (e.g., membrane material, pore size) are also well-known. Most studies performed to qualify an alternate filter or component can be completed before the alternate component is introduced into full-scale production (such as microbial ingress, extractables/leachables, compatibility, adsorption studies, and virus clearance studies, if applicable). Vendor studies supplemented by additional small-scale studies performed by companies enable the evaluation of essential performance characteristics before full-scale production. Changes that typically require health authority approval before use include changes to filters (final drug product or drug substance filtration, viral filtration, UF/DF) and to protein A affinity resin.
The intended qualification approach begins with a brief outline of the purpose of the step and change proposed. Next, a description of the characterization studies and supporting small-scale studies would be provided in a PACMP, which would be filed as a PAS.5 The PACMP would outline plans for confirmatory at-scale production runs and predefined acceptance criteria, which is aligned with the ICH Q12 guidance and recent FDA presentations.3, 5, 6 Third, confirmatory at-scale data would be submitted via the US ARMC. For older processes, process characterization and any impacts on product quality may be assessed using updated analytical methods, if applicable. Finally, a commitment may be provided to put the first post-change drug product lot (and drug substance lot, if the drug substance is liquid) on stability as an ad hoc lot, in addition to the typical annual stability measurements, based on the potential of the change to impact attributes that may be impacted by stability testing.
| Details | Approved Filter |
Proposed Filter Option 1 |
Proposed Filter Option 2 |
|---|---|---|---|
| Supplier | 1 | 2 | 3 |
| Membrane material |
Single- or dual-layer hydrophilized PVDF (or PES) |
Single- or dual-layer hydrophilized PVDF |
Single- or dual-layer hydrophilized PES |
| Nominal pore size |
0.22 or 0.2 µm | 0.22 or 0.2 µm | 0.22 or 0.2 µm |
| Sterilization method |
Autoclavable | Autoclavable or Gamma-irradiated |
Autoclavable or Gamma- irradiated |
| Filtration time | ≤ 72 hours | ≤72 hours | ≤ 72 hours |
| Filtration temperature |
2°C to 30°C | 2°C to 30°C | 2°C to 30°C |
| Maximum filtration pressure |
15 psig | 15 psig | 15 psig |
PVDF = polyvinylidene fluoride, PES = polyether sulfone
In addition to these changes (which are typically completed prior to approval), alternate sources for other product contact filters and single-use components may be reported via the ARMC or a CBE-0 before implementation, provided they have minimal or no impact to typical details provided in Module 3 of a Biologics License Application (BLA), based on risk assessments.
The accelerated filing proposal provided in this article could speed up the time to implementation of an alternate filter or resin by 4 to 6 months compared to a traditional filing, as shown in Figure 1. However, 12 months are typically required before implementation, even when using the accelerated filing. It is possible that in some cases where resin and filter supply risks pose an imminent threat to drug supply, more rapid use of alternative filters and resins will be required. In these cases, companies can consider filing small- and/or pilot-scale characterization data, risks assessments, prior knowledge, and vendor data as a CBE-30 before implementation at scale. Large-scale implementation would be managed through the product quality system and reported via the ARMC. Consideration would have to be given to established process conditions.
This approach would allow implementing alternative resins and filters within 6 months and would provide a rapid method of alleviating supply constraints; therefore, it would merit prior discussion between agencies and manufacturers. Four proposed data packages and submission strategies follow.
Alternative Bioburden and Sterile Filtration Filters
The filtration step is critical for aseptic manufacturing of parenteral drug products. Filter performance (i.e., microbiological control, no product impact) is determined by membrane material, pore size, and effective filtration area. The qualification and validation of alternate filters for bioburden reduction filtration and sterile processing steps may be necessary for aseptic processing of commercial antibodies.
| Characterization Study |
Proposed Filter Alternates |
Study Description |
|---|---|---|
| Filterability | 1, 2, 3 (Scale-down filter) |
Scale-down study using alternative filters would be used to confirm no practical change in filter ability of the product and confirm the appropriate filter size needed for currently validated batch sizes. |
| Product quality impact from filtration |
1, 2, 3 (Scaledown filter) |
Scale-down study would demonstrate no product quality impact due to filtration and contacting alternative filters for the maximum validated filtration time. |
| Initial surfactant/protein binding to filter |
1, 2, 3 (Scale-down filter) |
Surfactant and protein adsorption on alternative filters are expected to be similar to the approved filter due to the use of the same filter membrane material. Adsorption data would still be evaluated in separate, filter specific studies using scale-down filters. Data would be used to assess any changes in the strategy for ensuring acceptable product quality at the start of filtration (e.g., discarding or refiltering the initial filtrate). |
| Study | Proposed Filter Alternates |
Study Description |
|---|---|---|
| Microbial retention | 1, 2, 3 (Scale-down PVDF filter) |
This scale-down study would mimic the
|
| Filter leachables/ extractables |
1, 2, 3 | This study would be performed for alternative filter options using the sterile filtration method used during commercial manufacturing. |
The alternative filters could be sourced from the same or a different vendor, but otherwise would generally have similar pore sizes, a similar effective filtration area, and similar acceptable ranges of relevant filtration process parameters. A technical and risk assessment of the physical characteristics, material of contact, and performance and filterability data would be performed to demonstrate that the replacement filters meet all acceptance criteria of approved filters used for drug product manufacturing when operated within the acceptable range of process parameters. A comparison of the proposed filter options is shown in Table 1.
Proposed characterization
Tables 2 and 3 summarize the proposed process characterization and filter validation studies needed to support the use of alternative filters for bioburden reduction and sterile filtration steps.
The maximum filtration time that is established based on the media fill validation and the filter validation package would be applied to the new alternative filters. Additionally, companies would perform at least one at-scale process confirmation run per product incorporating the new alternative filter or filters to confirm no impact to the drug product manufacturing process and product quality attributes (e.g., via lot release or homogeneity of critical product quality attributes throughout the batch).
Compared to inline sterile filtration, bioburden reduction filtration is further away from the final product and, therefore, is less critical to product quality. If different filters for bioburden reduction filtration and sterile filtration are used during drug product manufacturing, the acceptability of the alternative filter options for bioburden reduction filtration can be assessed based on prior knowledge with limited product-specific characterization data. The alternative filters for sterile filtration are more critical (immediately before the filling operation) and require a more rigorous data package.
The microbial (bacterial) retention ability of a sterile filter is critical to ensure product sterility. Microbial retention validation, typically performed in a scale-down study with a long lead time (6 to 12 months), can be a major bottleneck to the submission of the data package. In this case, it is proposed that a manufacturing-scale study be performed at target conditions for initial submission with the microbial retention validation data package to be submitted later to justify process parameter acceptable ranges.
Proposed submission strategy
A PACMP as a PAS would be submitted. It would contain the description of the assessment for confirmatory testing at scale and provide the following data:
- Process characterization data from scale-down studies, as described in Table 2.
- Filter validation data, as described in Table 3.
Upon completion of the confirmatory at-scale testing, where the predefined acceptance criteria in the PACMP are met, companies would provide this data in a subsequent ARMC for the corresponding product to support the pre-filter and filter changes.
In the event that filter shortages only impact the bioburden reduction filter (not the final sterile filter), internal characterization would be performed (Tables 2 and 3), and the change would be reported in the ARMC. This approach is aligned with guidance presented recently by the FDA. Making changes to a drug substance bioburden reduction filter would involve a subset of the activities required for a drug product sterilizing filter and is described as follows.
Alternative Viral Filtration Filters
Multiple commercial molecule programs may be impacted by insufficient supply of viral filters (VF). The VF step is a filtration process designed to remove potential viruses through size exclusion. The VF step may be a two-stage filtration, with a pre-filter followed by a virus filter.
To address VF filter supply limitations across manufacturing, registration of an alternate pre-filter in addition to the approved pre-filter may be appropriate. The virus pre-filter from alternative vendors would be characterized and implemented at-scale. Comparison of currently approved and proposed VF pre-filters (Table 4) and filters (Table 5) are shown here.
Proposed submission strategy
A PACMP as a PAS would be submitted. It would contain the description of the assessment for confirmatory testing at scale and provide the following data:
- Lab-scale characterization data supporting the acceptance criteria for performance indicators and process parameters of the alternate pre-filter and filter. Characterization studies would include a demonstration of suitability via acceptable performance indicators. Qualification of a scale-down model may not be necessary or may be performed later using large-scale data for comparison. In addition, the studies would confirm the appropriate pre-filter and viral filter area needed for the viral filtration step and confirm no impact to product quality.
- The filter leachables and extractables would be assessed leveraging vendor data. The risk assessment would also be updated, if required.
- Viral clearance study data demonstrating effective virus removal. Use of a worst-case virus (mouse minute virus [MMV]) to assess virus clearance of the VF step for each product under challenging conditions (e.g., high volumetric loading) should be submitted to ensure viral clearance meets or exceeds a robust level reduction (4 log10) of small, non-enveloped viruses.
Viral clearance studies would demonstrate that the primary function of viral clearance is achieved. The lab-scale studies would confirm there is no product impact with use of the proposed alternate filters.
Upon implementation of the alternative filters into full-scale manufacturing, confirmatory testing would be performed on one batch at manufacturing scale. The at-scale data supporting the VF pre-filter and filter changes would likely include volumetric flux, volumetric throughput, integrity test value, and assurance that all release specifications and process controls are met. The data would be provided in a subsequent ARMC.
| Details | Approved Pre-Filter |
Proposed Pre-Filter Option 1 |
Proposed Pre-Filter Option 2 |
|---|---|---|---|
| Supplier | 1 | 2 | 3 |
| Filter media | Cellulose, inorganic filter aid |
PES | Nylon |
PES = polyethersulfone
| Details | Approved Filter | Proposed Filter Option 1 |
Proposed Filter Option 2 |
|---|---|---|---|
| Supplier | 1 | 2 | 3 |
| Filter media | PES | PES, PVDF, or cellulose | PES, PVDF, or cellulose |
| Nominal pore size |
≤20 nm | ≤20 nm (e.g., 15 nm, 20 nm) |
≤20 nm (e.g., 15 nm, 20 nm) |
PES = polyethersulfone, PVDF = polyvinylidene fluoride
Alternative Tangential Flow Filtration (UF/DF) Membrane
Multiple product programs could be impacted by insufficient supply of the UF/DF membranes. UF/DF steps are typically designed to buffer exchange product pools and to concentrate to the drug substance target concentration. The UF/DF filter retains the product protein and allows smaller salts and liquids to pass through. To address UF/DF filter supply limitations, registration of an alternate UF/DF filter in addition to the approved filter may be appropriate to mitigate filter supply risk.
A comparison of an approved and proposed UF/DF filter is shown in Table 6. A technical and risk assessment of the physical characteristics of the alternative filters should demonstrate that they are comparable or superior to the current approved UF/DF filter. Analysis and historical knowledge can be used to assess that no impact to product quality is expected.
Proposed submission strategy
Evidence for filter suitability and no product quality impact, which would include data from the filter manufacturer and data from scaled-down development studies, would be submitted as a PAS. The following would be provided:
- Lab-scale characterization data to support the acceptance criteria for performance indicators, process parameters, cleaning, and filter reuse. Characterization studies using a scale-down model and a demonstration of comparable properties via acceptable performance indicators would be submitted. In addition, the characterization study would confirm the appropriate filter size needed for the UF/DF step and confirm no impact to product quality.
- The filter leachables and extractables would be assessed leveraging vendor data. The risk assessment would also be updated, if required.
- Additional characterization data to support cleaning and reuse of membranes for the filter lifetime study, supported with at-scale data. It is possible that the cleaning method for the new filter will differ from the legacy cleaning. Measurements of attributes indicative of microbial control (bioburden and endotoxin) would be performed in addition to process performance indicators to confirm cleaning and use after storage.
| Details | Approved Filter | Proposed Filter Option 1 |
Proposed Filter Option 2 |
|---|---|---|---|
| Supplier | 1 | 2 | 3 |
| Filter media | Regenerated cellulose or PES |
Regenerated cellulose |
PES |
| Molecular weight cut-off |
30 kDa | 30 kDa or lower | 30 kDa or lower |
PES = polyethersulfone
| Details | Approved Protein A Resin |
Proposed Protein A Resin Option 1 |
Proposed Protein A Resin Option 2 |
|---|---|---|---|
| Supplier | 1 | 2 | 3 |
| Ligand | Recombinant (wild type) or engineered protein A |
Recombinant (engineered) protein A |
Recombinant (engineered) protein A |
| Backbone | Controlled pore glass or cross-linked agarose |
Cross-linked agarose | Cross-linked agarose |
Characterization data would ensure the UF/DF step meets the desired purpose of buffer exchange and/or concentration of the protein to prepare for drug substance formulation. The lab-scale cleaning studies would demonstrate no product impact with filter reuse.
Upon approval of the PAS, confirmatory at-scale data supporting the UF/DF filter change would be provided in a subsequent ARMC. Confirmatory at-scale filter cleaning and reuse data up to the maximum characterized number of uses would be provided as the manufacturing schedule allows through a future ARMC.
Alternate Protein A Affinity Chromatography Resin
Multiple product programs are likely to be impacted by insufficient supply of the protein A resin. To address protein A affinity resin supply limitations, registration of an alternate protein A resin in addition to the approved resin may be appropriate to mitigate supply risk.
Product-specific data and historical knowledge with other products (i.e., prior knowledge), vendor data, and risk assessments can be used to determine that no impact to product quality is expected when operated within the acceptable range of process parameters. A comparison of the proposed resin options is shown in Table 7.
Proposed submission strategy
A PACMP as a PAS would be submitted. It would contain the description of the assessment for confirmatory testing at scale and provide the following data:
- Data from characterization studies (including downstream process linkage) demonstrating comparability of product quality. This includes lab-scale characterization data to support the acceptable ranges for process parameters (including resin cleaning and reuse). Qualification of a scale-down model may not be necessary or may be performed later using large-scale data for comparison.
- The resin leachable and extractables would be assessed leveraging vendor data. The risk assessment would also be updated, if required. The leaching of protein A ligand would be measured as part of the characterization and at-scale studies using an assay appropriate for detection of the specific ligand would be performed.
- Additional characterization data to support cleaning and reuse of resin for the lifetime study, supported with at-scale data.
- Viral clearance study data demonstrating effective virus removal (if applicable).
Characterization data would ensure the protein A affinity chromatography step meets the desired purposes of the step: product capture, product concentration, and initial purification. The lab-scale cleaning and lifetime studies would demonstrate no product impact throughout the life of the resin.
Upon implementation of the alternative resin into full-scale manufacturing, confirmatory testing would be performed on one batch at manufacturing scale. The at-scale data supporting the resin change would likely include yield, purity, operating conditions, and assurance that all release specifications and process controls are met. Upon completion of the confirmatory at-scale testing where the predefined acceptance criteria in the PACMP are met, companies would provide this data in a subsequent ARMC for the corresponding product to support the resin change.
Confirmatory at-scale resin cleaning and reuse data up to the maximum characterized number of uses would be provided as the manufacturing schedule allows through a future ARMC. This approach may also be employed for other chromatography steps if the alternate resin can be implemented with minimal impact to process parameters and the downstream process.3
Other Alternative Filters and Single-Use Components
To address potential supply constraints throughout manufacturing, an evaluation of alternatives for several product-contacting filters would also be performed. The key information and data needed to assess the suitability of these filters would be evaluated before use in production.
Example 1: Depth filters
Proposed change: An alternative depth filter for direct cell capture or post-centrifuge harvest operations would be added. All registered process parameters and performance indicators would remain within the currently approved characterized range.
Characterization studies would include a combination of existing vendor and internal lab-scale studies. To establish equivalency, an assessment would be performed against the following key parameters:
- Filter capacity
- Permeability
- Yield
- Extractables/leachables
- Impact to levels of process-related impurities
Based on the analysis and available small- and large-scale data, the alternative filter would be used interchangeably with the approved filter in depth filtration operations.
Module 3 impact: An update to the list of raw materials may be required. Conclusion: Successful completion of the technical assessment, risk assessment, and characterization studies before implementation would ensure that this change has minimal potential to impact product quality, and this change would be summarized in the ARMC.
Example 2: Product Contact Bioburden Reduction Filters
Proposed change: An alternate source for the product contact bioburden reduction filters, typically used before and after each unit operation throughout the drug substance process, would be added.
Evaluation to be completed before implementation: The performances of two 0.2-micron filters would be compared based on filter sizing studies of several in-process streams. The comparison of the two filters for in-process and drug substance final filtration would be experimentally assessed against the following key parameters: filter capacity and permeability.
Vendor data for the following attributes would be assessed and considered during a risk assessment: Retention rating; extractables/leachables; and sterilization method.
The extent of the comparison could vary depending on the perceived level of risk. Filters upstream in the process, followed by several bind-elute separation steps may present a lower risk than a filter used for a liquid drug substance. Based on the analysis and available data, the alternative filter could be used interchangeably with the approved filter in UF/DF recovery filtration and drug substance final filtration. Filter sizing would depend on the capacity as determined by the applicable small-scale model.
Module 3 impact: An update to the list of raw materials may be required. Conclusion: Successful completion of the characterization studies before implementation would ensure this change has minimal potential to impact product quality, and this change would be summarized in the ARMC.
Conclusion
To mitigate COVID-related or similar supply challenges, a strategy is proposed herein to generate a robust characterization package designed to demonstrate an alternative protein A affinity chromatography resin, filter, or component have low risk of product quality impact and submit this data/information as a PAS with a PACMP, and, if needed, request an expedited review. In addition to characterization data, confirmatory at-scale data would be proposed in the initial PAS and a commitment would be made to provide any at-scale product quality and/or validation data in the ARMC.
For new BLA submissions (non-COVID therapies), sponsors might consider describing one or more of the alternate filter or resin strategies as a PACMP. Lab-scale characterization data in the BLA could be sufficient to support the proposed full-scale testing plan and then reporting with the ARMC, without the need for a PAS.
Although supply chain shortages for filter components are of particular concern in the US, the pandemic has highlighted the interconnected nature of global manufacturing and supply chains in the biotechnology industry. For manufacturers that have products registered in many markets worldwide, the challenges with long and uncertain delivery times for essential components such as filters and critical raw materials are exacerbated by the complex regulatory processes already inherent in the management of postapproval CMC changes.7
For a single change that requires prior approval, the divergent data requirements, staggered submission and bundling strategies, and spread-out approval timelines in the different markets often mean that implementation takes several years. During this time, the manufacturer would be required to manage pre- and post-change product tightly to maintain compliance, often resulting in duplication of efforts and potential stockouts in cases of shortages of critical materials. It would thus be desirable to apply the regulatory strategy approaches described in this article for the FDA to other regulatory submissions.
With the endorsement of ICH Q12 in November 2019, and its ongoing implementation in the ICH member countries, this becomes a feasible global regulatory strategy.3, 4, 5 PACMPs are typically among the first ICH Q12 tools to be used once the individual regulatory frameworks have been adapted accordingly and are already accepted in several other countries. Thus, it should be possible to gain regulatory agreement via a PACMP (which would include laboratory-scale characterization data, risk- and science-based justifications, and appropriate acceptance criteria) that at-scale data could subsequently be submitted using a “notification low” category as per the harmonized ICH Q12 guideline. In turn, shortening the approval timelines globally will streamline the postapproval change management processes and mitigate the risk of drug shortages.





