Cleaning Validation Program Maintenance in a Process Life-Cycle Model

The process life-cycle model, as discussed in the US FDA guidance on process validation, is a significant change in how we view validation.
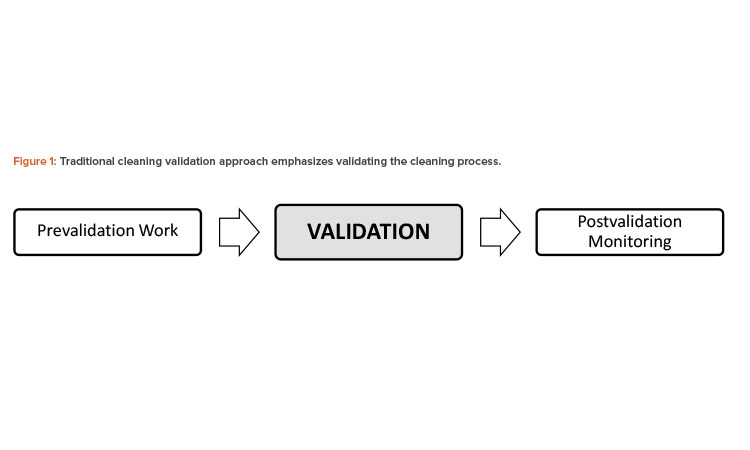
Over the past few decades, various cleaning validation guidance documents have provided the industry with insight on how to comply with individual country regulations.2, 3, 4 These guidance documents primarily focus on general validation aspects (see Figure 1). Although the prevalidation design phase and postvalidation monitoring stages were factored into the process, they are not explicitly indicated or emphasized in the regulatory guides. Today, this guidance is referred to as the “traditional cleaning validation approach.”
By building robust scientific knowledge before validation, the design phase is the base that supports the decisions made in the process. As presented in the 2011 US FDA process validation guidance,1 the design phase calls for up-front work and use of modern tools such as risk evaluation (e.g., design of experiments, risk ranking), bench- or pilot-scale experiments, and novel equipment and instrument technology.5 In contrast, many validated clean-ing validation programs under the traditional approach have minimal documented work to fully support, for example, the best selection of critical parameters or methods to determine the impact of those parameters on cleanability.
For GMP manufacturing processes where new cleaning procedures (or improved ones) are being considered, applying a three-stage process life-cycle validation approach is more feasible and justifiable than the traditional approach. GMP manufacturers must ensure that the site is equipped with the necessary resources and technology early in the development of the new cleaning procedure. This enables the manufacturer to successfully complete the design phase, which helps streamline the qualification and, subsequently, the monitoring stage of the product life-cycle model.6
However, there remains an underlying question about current manufacturing processes with cleaning procedures validated under the “traditional” approach (e.g., legacy drug products): Must companies revalidate cleaning procedures starting from scratch to perfectly comply with the three-stage process life-cycle model?
For now, systems must be in place to supplement any validated cleaning program regardless of the extent of prevalidation work. GMP manufacturers must at least assess the risk of the current cleaning procedure and provide assurance that it performs as validated and remains in a state of control for the life of the product(s) being manufactured. Failure to establish an adequate ongoing monitoring program, or at least a periodic revalidation program, is likely to result in sanctions from health authorities.7 Only time will tell whether the local and global regulatory expectations will change in the future.
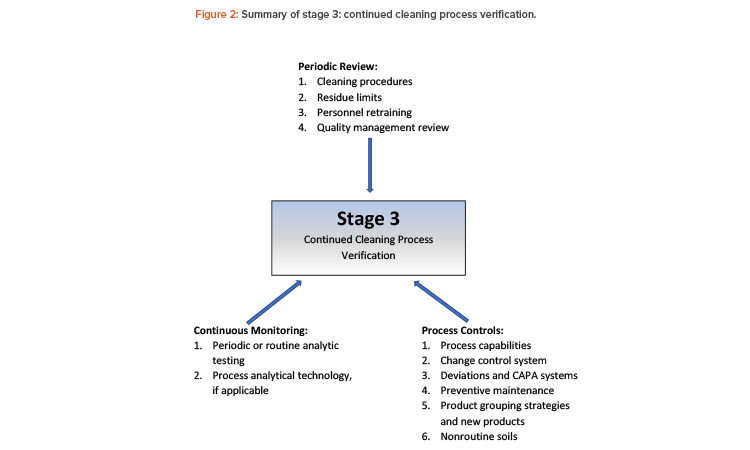
The system to evaluate the current cleaning process’s performance may include, for example, a collective assessment of cleaning-related data to detect undesired variability. This evaluation is equivalent to stage 3, continued process verification (or ongoing process verification, as it is called in the European Union).8
Figure 2 presents recommended elements to maintain validated cleaning procedures as part of a process life-cycle approach. Any number of these elements may be taken into consideration for different cleaning scenarios, and the selected elements must be established in a procedure, protocol, or master plan.
Product Grouping
Product grouping is a popular cleaning validation strategy used in multiproduct facilities. Products manufactured on the same equipment can be grouped together if the cleaning procedure is proven effective for cleaning the hardest-to-clean product in the group down to the acceptable residual limits of the most toxic product in the group. Other approaches include selecting a worst-case representative product based on a point risk-ranking system. Grouping is generally based on three aspects:
- Solubility of the active ingredient
- Cleanability of the process residue
- Toxicity of the active ingredient
In some cleaning procedures that were validated years ago, selection of the worst-case product is based solely on solubility data or solubility data combined with anecdotal evidence. This approach may trigger questions during an agency inspection about the validity of the worst-case selection.
A simple example can be used to illustrate the issue with using solubility data alone. One teaspoon of sugar poured into a cup of water at ambient temperature with mild stirring takes a few seconds to dissolve completely. However, if one teaspoon of sugar is poured onto a hot stainless steel coupon, melts, and then cools down, dipping the coupon in water at ambient temperature for a few seconds is unlikely to remove the sugar residue. In other words, the basic solubility information about sugar in water is insufficient to assess cleanability. Cleanability also takes into consideration the sur-face-residue interaction (such as residue conditions and the surface type) and how cleaning agents or cleaning mechanisms break that interaction.9 Solubility is often limited to the active ingredient and may not be representative of the entire process soil, especially if cleaning is performed using a cleaning agent other than water. For these reasons, grouping strategies lacking scientific data to support cleanability must be reassessed to provide better justifications in the selection of worst-case soils.
Cleanability of the process soils can be based on documented pilot plant or laboratory coupon testing. In addition to supporting the current worst-case selection, testing data are also important when introducing a new product into the same manufacturing train. Coupon studies can compare cleanability between the validated worst-case soil with new soil(s), along with an evaluation of the new soil’s toxicity. As shown in Figure 3, coupon testing can include coating a stainless steel coupon, or representative substrate, with the new soil and conditioning the coupon for a specified time and temperature.10 Once the coupon is conditioned, it can be cleaned using the same cleaning method applied for the current worse case.
Coupon studies can help confirm that the current cleaning process is effective for the new residue or determine that the new residue may be considered a new worst case. For example, when combined with a toxicological risk assessment, a residue acceptance limit greater than the currently validated limits may be used to show that the new residue is less toxic and to justify that a new cleaning validation is not required at the time. Alternatively, if the new residue’s acceptance limit is lower than the currently validated limits, a new cleaning validation may be necessary.

Review of Acceptable Residue Limits
Defining acceptance criteria remains perhaps the most challenging aspect of a cleaning validation program.
- How “clean” is the surface considered clean?
- How can an acceptable carryover limit from one product to another be established?
- How can surface cleanliness be demonstrated?
Historically, the commonly used method for determining residue limits is based on the Fourman and Mullen approach, also known as therapeutic dose–based calculation.11 In addition to a visually clean surface, this approach uses the more stringent of the following two criteria:
- Any active ingredient is less than 10 parts per million (ppm) in any subsequent product.
- Any active ingredient in a subsequent product is less than 1/1,000 of the minimum daily dose of the active ingredient in the maximum daily dose of the subsequent product.
Several articles have described procedures and reported average visual residual limits based on residues, surfaces, and other factors. 12, 13
Many pharmaceutical companies continue to support the dose-based calculation. However, recent industry publications and regulatory changes affecting primarily European countries are leading the way to a different approach, known as the health-based calculation.14, 15 Manufacturers may wish to evaluate and compare different approaches to residue limits calculation to determine which best fits cGMP requirements, corporate policies, and site objectives. Reviewing residue limits periodically to assess conformance with industry trends helps companies ensure that the validated limits are well within the market requirements where the drugs products are sold.
Cleaning Data Monitoring and Trending
As suggested in the FDA process validation guidance, to accomplish continuous assurance, the manufacturer must have a system (or systems) to detect unplanned departures from the validated process.1 An ongoing program to collect and analyze product and process data that relate to cleaning acceptance criteria must be established. The data should be statistically trended and reviewed by a statistician or cleaning subject matter expert.
Routine or periodic sampling must be specified in the cleaning procedure and recorded. The type of sampling, number of samples, sampling frequency, and analytical tests may vary per cleaning method. The routine or periodic sampling plan has a smaller number of sampling points than the validation sampling plan based on the results of the validation study and risk assessment. Routine sampling must be easily collected and tested after each cleaning execution. Technologies such as conductivity probes employed in automated clean-in-place systems are suitable for routine sampling. Periodic sampling may be considered for manual cleaning applications at some defined yearly frequency.
The routine or periodic sampling plan must allow the manufacturer to monitor critical cleaning attributes while minimally affecting the cleaning turnaround time. For example, specific analytical methods such as high-performance liquid chromatography (HPLC) are preferred for validation purposes, whereas nonspecific methods such as conductivity, titration, or total organic carbon (TOC) may be more suitable for routine use due to their fast response times. Similarly, rinse sampling may be selected over swab sampling for routine or periodic analysis because the swab sampling is the more invasive and time-consuming approach.
Process capability compares the output of a process to the specification limits by using capability indices. The comparison is made by forming the ratio of the spread between process specifications and the spread of process values, as measured by three or six times the process standard deviation units.16 The capability index (Cpk) is a value between 1.25 and 2.00 that represents the ultimate potential for a process to produce an output within specification. More information on process capability can be found elsewhere.16 In the case of cleaning validation, one-sided only, upper specification limit (USL) is often used to calculate Cpk as follows:
$$\begin{align} Cpk & = \frac{USL - µ}{3\sigma}\end{align} $$
where µ is the average of the measurements and \( \sigma\) is the standard deviation of the measurements.
Preventive Maintenance
Equipment and instruments employed in the cleaning procedure must undergo preventive maintenance on a regular schedule, which should be set up in advance for all critical equipment and instruments. A combination of equipment manufacturer recommendations, mechanical experience, usage characteristics, and substrate compatibility with cleaning agents can be used to assess the equipment’s risk of failure or deterioration and determine the frequency of maintenance. Preventive maintenance should include a calibration procedure for measurement devices such as weight scales, thermometers, flow cells, conductivity and pH probes, and other testing equipment used in the cleaning process.
Preventive maintenance in the cleaning program must address potential risk factors such as surface abnormalities. Discolored or damaged surfaces should be noted during routine visual inspection and scheduled surface inspections. Procedures should be in place to rate the severity of the abnormality and determine the corrective action, if needed. Periodic checks for worn gaskets, O-rings, dead leg orientation, sampling ports, and valves are also recommended to mitigate the risk of substrate deterioration that may result in batch contamination. Table 1 lists several preventive maintenance issues to consider in cleaning validation.
Cleaning Nonroutine Soils
The following are some examples of soils that are not routinely considered in a cleaning validation study because they generally occur in specific circumstances and are often not fully understood until they are investigated. In recent years, various industries have reported having issues of this nature. Nonroutine soils should be investigated, assessed, and remediated to mitigate their impact on the current validated procedure and, hence, product quality.
| Items | Aspect to consider |
|---|---|
| Valves and sampling ports |
|
| Dead legs |
|
| Spray devices |
|
| Piping and vessels |
|
| Pumps |
|
| Flexible hoses |
|
| Automatic valves, flow meters and other measuring devices, field test instruments (e.g., passivation test kit) |
|
| Water and gaseous utilities |
|
Rouge
Rouging can occur when stainless steel water generation systems, process tanks, and pipeline systems are routinely exposed to corrosive solutions. The US FDA has stated in at least one warning letter that corrosion is unacceptable in direct-contact pharmaceutical systems.17 Rouge on product contact surfaces creates an environment for process residues and microbes to tenaciously adhere to the rouged area, causing it to become more difficult to clean and disinfect.18, 19
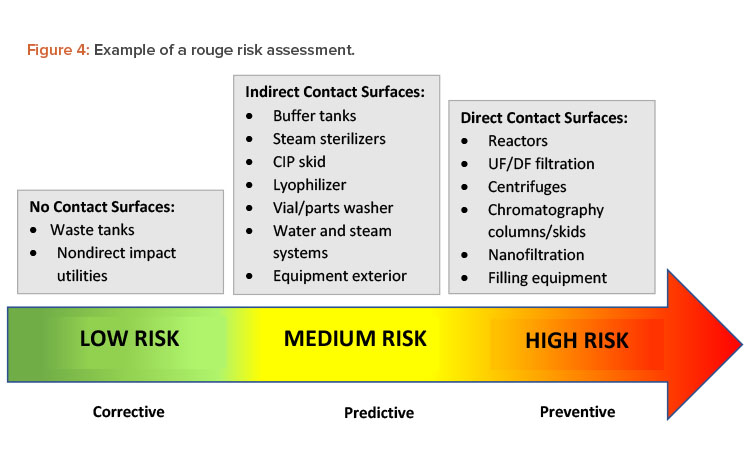
Some manufacturers use treatments to prevent rouge from happening in the first place. Other companies wait until rouge has been detected or has affected production to take corrective action. If a process or surface condition is known to cause corrosion that will at some point affect direct product contact surfaces, the manufacturer should try to prevent that corrosion from occurring. Figure 4 illustrates a risk assessment for rouge. When choosing the best strategy to address this risk, it is important to review the potential impact on patients, products, personnel, and equipment.20
An effective procedure for maintaining stainless steel surfaces in a passivated state and preventing corrosion requires a careful balance of several factors, including:
- Ability to remove any exogenous particles, including rouge
- Equipment and system constraints (e.g., temperatures, substrate compatibility, flow rates)
- Minimal or no surface damage caused by aggressive chemistries
- Personnel safety and disposal concerns
- Adherence to industry standards (e.g., ASTM A967,21 ASME BPE22)
- Concerns related to the use of chemicals that are not part of the validated cleaning process
Biofilms
A standard practice in cleaning validation studies is to consider intrinsic sources of bioburden, such as those introduced by raw materials. Cleaning procedures must be designed to be effective against both chemical and intrinsic microbial residues.
Cleaning procedures must also address extrinsic sources of microbial contamination in batches and/or equipment. Extrinsic contaminants can enter a system via air, liquid, or surface contact. Examples are gram-positive bacterial contamination resulting from poor gowning practices, fungal spore contamination from open process containers, gram-negative bacteria from process water, or spore-forming microbes from contaminated raw materials.23
Biofilms are associated mostly with gram-negative bacteria in process systems or utilities, but they can also involve other harmful microorganisms. Gram-negative microbes contain endotoxins, primarily in the cell membrane, which can build up on equipment surfaces and eventually lead to product failure. Biofilms are also more resistant to destruction than vegetative organisms. For these reasons, biofilm contamination is a serious concern for medical implant, parenteral, ophthalmic, and other GMP products.
When biofilms or endotoxins are present, the strategy required to remove the residue effectively may differ from the validated cleaning procedure. At times, this strategy is more aggressive than the validated cleaning procedure and must be combined with a thorough inspection of the equipment’s sanitary design to reduce the risk of microbial contamination reoccurrence. Published studies evaluated the inactivation of Bacillus cereus biofilm and recommended using a disinfectant with and without precleaning with a formulated alkaline cleaning agent.24, 25
Air-Liquid Interface Residue
Common buffers used in pharmaceutical and biopharmaceutical manufacturing processes are generally cleaned with water only, a strategy based on solubility data. However, trace levels of substances present in raw materials such as slip agents and particles from incompatible plastics and elastomers used in gaskets and tubing can migrate to blending and storage tanks walls. These trace substances are hydrophobic and can adhere tenaciously to the sidewalls, causing an air-liquid interface (ALI) buildup (or ring) over time.
Identifying the ALI ring components is the first step in determining the ring’s origin. Laboratory studies have shown to be effective as a starting point for choosing the optimal course of action,26 which might involve any of the following:
- Proving that a maintenance cleaning procedure cleans the equipment and either prevents the ring from forming or removes the ring once it is visible
- Modifying a cleaning procedure to clean the ALI residue before the residue is visible
- Identifying the source of the trace material and trying to eliminate it from the raw material through a corrective and preventive action (CAPA) plan
Common cleaning approaches include using a formulated alkaline cleaning agent at elevated temperatures, often with a detergent additive to increase the surfactant level with or without hydrogen peroxide.
| Changes to | May Impact | Actions to Consider |
|---|---|---|
| Detergent components | Ability to clean soils, residue limits for the cleaning agent, cleaning agent rinsability |
Coupon testing, toxicity reevaluation |
| Cleaning parameters (time, action, chemicals, and Temperature) | Ability to clean soils | Coupon testing |
| Analytical method | Detectability and quantification of residue(s) | Analytical method validation (e.g., limit of detection, limit of quantitation) |
| Equipment design | Surface coverage, drainability, change over time, substrate compatibility | Spray device qualification, coupon testing |
| Personnel | Training and necessary level of experience | Classroom and on-the-job training |
| Dirty hold time | Ability to clean soils, levels of bioburden | Coupon testing, microbial tests |
| Cleaning hold time | Extraneous matter, bioburden | Microbial tests |
| Length of campaign | Ability to clean soils | Coupon testing, full-scale qualification run |
| New product in the same train | Grouping strategy | Coupon testing, toxicity reevaluation |
| Manufacturing steps or operational parameters | Soil condition and cleanability | Coupon testing, full-scale qualification run |
| Batch components or raw material sources | Cleanability, bioburden | Coupon testing, toxicity reevaluation, microbial tests |
Personnel Retraining
It is a standard practice, and a regulatory requirement in some countries, for pharmaceutical companies to periodically review their procedures on a pre-established basis according to company policies—usually every two to three years. The review may involve editorial changes to improve the clarity of operator instructions, but these changes must not significantly alter or change the current validated procedure. A personnel retraining session should be part of the periodic procedure review when procedures are changed. Even when procedural changes are not made, personnel should be periodically retrained in cleaning. As a rule, the more reliant the procedure is on human intervention, the greater the frequency of training should be. Most companies conduct retraining every 3 to 12 months for manual cleaning applications, which have inherent operator-to-operator variability, and schedule retraining for fully automated training every two to three years.
Change Control Procedures and Deviations
When manufacturers need to propose planned or unplanned changes to routine operations, these proposed actions may have an impact on the cleaning process. There are cases in which evaluating the impact of the change on cleaning may include laboratory coupon testing, as previously discussed. Therefore, validated cleaning procedures must be included in the change control management system, which ensures that any proposed changes are evaluated fully for their impact on the validated state of the procedure.
Change control systems may affect all or part of the cleaning process in multiple ways, as illustrated in Table 2. This table is not an all-inclusive list but provides examples of changes and their potential impact on cleaning procedures. The company’s change control procedure must include a section for the evaluation of the impact of cleaning validation by a designated subject matter expert (SME) within the organization.
The cleaning SME should approve changes before they are implemented. For major proposed changes, the change control management system should coordinate an assessment of the changes and determine whether new validation is required. When new validation is determined to be necessary, the three-stage life-cycle validation model should be implemented.
Manufacturers should fully investigate out-of-specification (OOS) cleaning results (e.g., from visual inspection or analytical data) and deviations from a validated cleaning procedure, per local procedures, and address the issues appropriately. The cleaning SME should provide the initial assessment and also determine the next course of CAPAs when the investigation is completed. In the case of an OOS event, the equipment should not be used for the next product until the equipment has been cleaned, met all cleanliness acceptance criteria, and been released by the quality unit.
CAPAs for a cleaning issue should be based on the results of a risk assessment. The cleaning SME should be responsible for ensuring that the root cause analysis and proposed corrections are appropriate to address the cleaning issue. Sources leading to initiation of a CAPA related to cleaning may include (but are not limited to):
- Quality management review
- Product contamination incidents
- Audit inspections
- Data trending from monitoring
- Customer complaints
A formal review of the cleaning program should be conducted at least annually and may be conducted as part of the required product annual review. The quality unit should document the formal review. At a minimum, the annual review should include a collective summary of cleaning-related deviations, CAPAs, periodic monitoring, and change controls that have an impact on cleaning validation.
Conclusion
The traditional cleaning validation approach has been used for over 30 years to validate cleaning within cGMP manufacturing. The three-stage life-cycle approach adds emphasis from validation to design and monitoring of the cleaning process. Companies should consider establishing a monitoring stage in a cleaning program to be feasible and necessary regardless of the validation approach taken. Systems must be in place to supplement any validated cleaning program regardless of the extent of prevalidation work. Failure to establish an adequate ongoing monitoring program is likely to result in sanctions from health authorities.



