Moving From Cleanroom to Isolation Technology for ATMPs

Advanced therapy medicinal products (ATMPs) pose specific manufacturing challenges beyond those typically addressed by pharmaceutical chemistry. Often in current ATMP applications, a change in approach is introduced at some point in the development process out of convenience or necessity, which then results in a change in technology. This article analyzes the possibility of transferring a cell and gene therapy (C>) process from the cleanroom approach to an environment based on isolation technology or, in other words, of moving the process from an open space manipulation to a closed and segregated space concept.
The development of C> applications raises several questions about the options available for production environments. Although many C> processes originated from cleanroom technology, it is reasonable to assume that the time has come to use more efficient production methods.
The aim is to improve quality assurance and efficiency in production of C> products, thus generating an impetus to contain costs while improving application possibilities and availability for eventual users. Isolation technology represents an improvement in this field and has proved effective in many other pharmaceutical chemistry applications. Transitioning a C> process from a staffed cleanroom—with operators in sterile scrubs working in Grade B environments under biosafety cabinets (BSCs)—to a closed and segregated environment—with operators working from the outside—has challenged operators and process engineers to find the best method to reproduce an established and validated process. In this article, we summarize the main differences between the two approaches, discuss the main problems that emerged, and provide several possible solutions for a smooth and faultless process transition.
ATMP Applications
Often in current ATMP applications, at some point in the development process, a change in approach is introduced for reasons of convenience or necessity, which then results in a change in technology. Many of the current in-development processes at the preclinical stage started in an academic environment, dealing with open processes in BSC, with highly operator-centric manufacturing. When the progresses manifest the need of a transition to early clinical production, we need to look at upgrading to a more robust, technology-based, closed platform.1
In the past, the open cleanroom approach was dominant and prevalent, but the production of ATMPs requires different technologies, isolation technology in particular. It has been encouraged and recommended by authorities1 because it offers better quality assurance in production (in particular, stability of the environmental conditions, lower levels of contamination, and availability of environmental data) and because it reduces costs.2, 3, 4, 5
In this case study, we analyzed the possibility of transferring a C> process from the cleanroom approach to an environment based on isolation technology. In moving the process from an open space manipulation to a closed and segregated space concept, it became clear that it is not possible to simply replicate the whole process. Instead, the task required, depending on the different situations, adaptation of the procedures and tools.
We will analyze this process change here, offering the reader the considerations we encountered and the solutions adopted. In some cases, these solutions consist of having a replacement procedure. In other cases, we needed to develop new technologies to facilitate adapting the open-environment procedure into one with comparable performance in the segregated approach.
As extensively reported in several publications,1, 6, 8, 10 ATMPs pose specific manufacturing challenges, going beyond those typically addressed by pharmaceutical chemistry. While out of scope of this article, these differences include working with living cells, which means dealing with contamination along with many other factors, which we file under “cell characterization.”
Finally, we must ensure that the preparation we deliver to patients has the right potency, e.g., when correctly administered, it has the intended result. Because we are dealing with living cells, this can be affected by different factors, including manufacturing, delivery, and transportation. Given the relatively short life of these products, C>s require tight coordination between physicians and manufacturers, as products are often manufactured for individual patients, and changes in timing can impact physicians’ treatment decisions.6, 7, 8
We know from experience that the development process of C> products is often not complete in the initial phases of the project and is subject to several improvements, modifications, and integrations while in process. Therefore we need to also adapt our manufacturing processes to the results obtained during the development itself.
The 5- to 10-year future of the product should also be considered. When production starts, capacity consists of a handful of patients per year. But if the therapy is successful, production capacity must be increased to meet demand.
According to Walters-Nelson, “The biggest issue we tend to see as you start to get into commercial manufacture relates to the increase in patient population.”1
This increase leads to a demand to either scale up, if the process is autologous, or scale out, if the process is allogeneic. Other concerns include how to ensure scalability of your process is not introducing a linear increase of the workforce, or of the footprint of the facility. As noted by Walters-Nelson, “If you’re going to a 1,000, 2,000 or 5,000 patients per year facility and you are looking at linear expansion, you don’t want to have to grow your facility five-, six-, ten-fold to accommodate demand. You must think about how to do manufacturing in a more efficient manner.”1
Project Background
Most considerations reported here are part of work completed assisting a customer with an existing laboratory working in a Grade B environment who wanted to extend the operations but move to an approach based on isolators and a Grade C cleanroom. According to regulations,1, 11 isolators can run in Grade D cleanrooms, but the customer already had a Grade C cleanroom available, so there was no need to perform additional work to downgrade the classification.
The company is a private organization working with umbilical cord blood collection and is classified as a cord blood bank. It follows FACT standards9 and is one of a limited number of companies to have this accreditation. Being in Switzerland, it is authorized for medical practice under Swissmedic, the Swiss National Certification Agency. The authorization also covers mesenchymal stromal cells (MSC) manufacturing.
Technical Challenges and Choices
In this case study, we relate the transition from the open environment to a closed environment. In the open environment, after several steps of clothing changes with increasing cleanliness, sterile-gowned operators come to a Grade B environment, in an open space, and directly handle products under BSC. In the closed environment, isolators are placed in a Grade C/D background, and operators never come in direct contact with products, which are kept inside the closed Grade A segregated environment and manipulated with the use of suitable glove ports.
The regulatory framework is well known; all national regulatory bodies have issued recommendations on the manufacturing of ATMPs.11, 12 In Europe, the reference documents are EudraLex–Volume 4–Good Manufacturing Practice (GMP) Guidelines11 together with EU regulations concerning Pharmaceutical Aseptic Manufacturing,1 which Swissmedic is adopting in Switzerland.
The customer in our case study wanted to change their approach to reduce risk of contamination and cross contamination, achieve flexibility and procedural simplification (e.g., easier access, no more sterile gowning, simplified validation, fast and reliable cleaning), and save money on operational expenses.4, 5 With moving to a closed technology, several operations and construction details had to be adapted or newly introduced to match the different requirements of this new approach.
Finally, the customer had expectations for final product quality and plant throughput with respect to the space occupied (sometimes referred as batches per year per sqm of surface occupied), with the possibility that operations could be expanded or modified in the future. Further, the product cost had to be kept as low as possible to facilitate patient access and to support process commercialization.10
Analysis
To analyze the impact of the proposed technology on the production process, we focus our attention on aspects concerned with the following:
- Gloves
- Material management
- Sampling, product delivery, and waste management
- Innovation
- Automation
Gloves
Gloves are a critical point to consider when switching between an open environment and closed environment. Isolator gloves are often cumbersome: they are thicker than cleanroom gloves currently used, users claim to experience a loss of sensitivity, users worry isolator gloves could slow activities, etc. The thickness of isolator gloves is necessary because they must be able to withstand regular hydrogen peroxide decontamination runs: they must be resistant to the biocide agent and thick enough to retain their resistance to punctures and tears through successive decontaminations. Typical isolator gloves thicknesses are 0.4 mm and 0.6 mm. In our experience in the pharmaceutical industry, we find that chlorosulfonated polyethylene (CSM) gloves with 0.4 mm thickness achieve the best compromise between sufficient flexibility and dexterity and resistance to vapor phase hydrogen peroxide (VPHP) decontamination.
Our experience verifies the claim that there is reduced sensitivity when using the isolator gloves; however, this tends to diminish and then disappear as operators are trained with their use. A typical training session involves the repetitive use of commonly used devices in successive sessions and involving all operations. Initial training sessions can be simulated without the products, freeing operators from responsibility and letting them concentrate on achieving the right dexterity.
To overcome the inevitable differences between the sterile surgical-type glove used in grade B cleanrooms and the 0.4-mm-thick isolator glove, we set up a mock training session with the following operations:
- Picking up and unscrewing the cap of a 2 ml cryovial (Corning product number 430659)
- Pipetting the content
- Screwing on the cap of the cryovial
- Unscrewing the cap of a prefilled Falcon 50 (REF 352098)
- Diluting the pipetted content in the Falcon 50
- Screwing on the cap of the Falcon 50
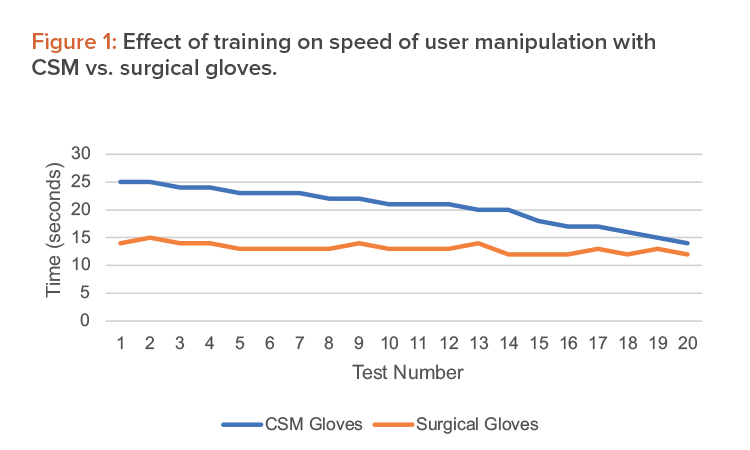
Average results from the training activities are shown in Figure 1.
From this, we verify that although there may be some difficulties using the CSM gloves in the beginning, after a set training time, operations run incomparable time and users obtain a comparable dexterity. We have estimated this training time in the operating setup as one week, including repeated simulation of the single operations as well as full process runs.
| Material | Packing | Physical Condition |
|---|---|---|
| Pipettes and pipette tips | Plastic wrapping | Dry |
| Flasks | Plastic wrapping | Dry |
| Bottles of cell culture media | Unwrapped | Dry/room temperature |
| Tubes | Wrapped | Dry |
| Cryovials (empty) | Plastic wrapping | Dry |
| Cryovials (filled) | Unwrapped | Wet/frozen |
| Microscope slides | Packed | Dry |
| IV Bag | Unpacked | Room temperature or 4°C |
Material Management
Different materials are needed to carry out ATMP processes. Table 1 shows a sample, not exhaustive, list. These materials can be classified according to their nature and the physical condition they arrive in. As shown in the table, materials may be wrapped (and sometimes double-wrapped), unwrapped, at room temperature, frozen, etc.
Materials are introduced to a Grade A cleanroom through a succession of material airlocks and a series of decontamination passages using air flushes and suitable disinfectant agents. Typical decontamination while working in isolators is to use an airlock with a decontamination cycle based on hydrogen peroxide vapors. Materials are introduced in the airlock through the front door and the cycle starts. When finished, the interlock between the main chamber and the airlock is released, and the materials can be passed into the processing chamber. A typical decontamination run can take 25 to 45 minutes, depending on the internal load of the chamber. Hydrogen peroxide cycles follow predetermined recipes, set during performance qualification of the equipment.
Due to the different nature of the materials and the different conditions under which the materials are presented, in an isolator environment, an appropriate differentiated strategy may need to be used for material introduction, combining hydrogen peroxide decontamination and safe manual cleaning. In the first step, in a laminar airflow pre-chamber equipped with gloves, laminar flow is used to clean the material and the operator performs a suitable spraying with a sporicidal in the first run. In a second step, the operator passes the material to the airlock (center), waits for reestablishment of Grade A–level cleanliness (with a laminar airflow speed of 0.45m/s, we have in that pre-chamber a safe recovery time of a couple of minutes, as checked with particle counter during validation), and then the material is passed to the processing chamber. All these operations have been validated by the validation team, during system acceptance, including correct pressure cascades (main chamber > pre-chamber > laminar airflow).
Whether working in a cleanroom or in an isolator, a clear supply plan for consumables and ancillary materials is mandatory. When working in a cleanroom, the space allows keeping significant stock, so you can pick up items requested by the process. A typical way to solve this issue in an isolator environment is to provide a storage area where basic working stock is located. This will help avoid too many replenishments during the course of the process, which mitigates any risk of external contamination.
Sampling, Product Delivery, and Waste
A common activity when running an ATMP process involves collecting samples for in-process controls. Depending on the nature of the final product, a similar activity also has to be run at the end of the procedure for product delivery. Given the different nature of the products that could be developed as ATMPs, blood bags, vials, or other standard or customized containers for tissues all deserve a different approach for packaging and product exit. In the cleanroom, the typical way of realizing the output of finished products is to reverse the process of product input, but the isolator’s compact working area provides the possibility to separate them and maintain a unidirectional flow of materials and process.
Sampling
Sampling is an essential operation to in process controls (IPC). It is very similar to product exit, except that sample aliquots are small quantities often contained in vials or in specific instruments cartridges aseptically filled inside the isolator.
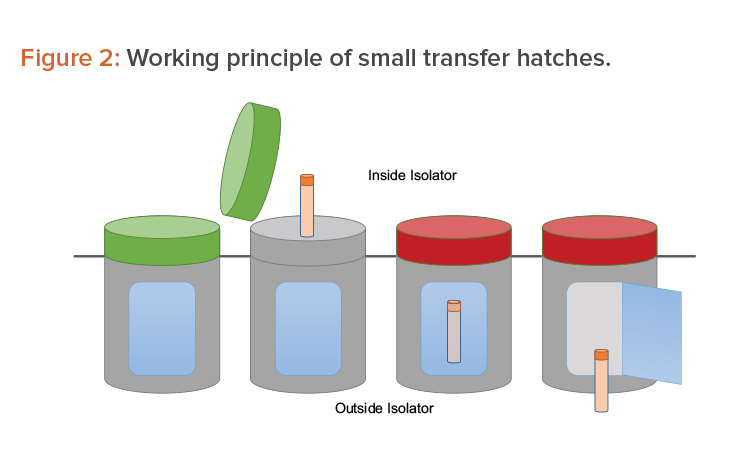
Many options are available to accomplish the operations mentioned previously. One option is to use a series of small, single-use transfer hatches. The hatches face the Grade A environment before being used. Once filled with sample, they are locked by a sealing gasket (a red light indicates the hatch cannot be opened). A second opening—interlocked with the first one—faces the Grade D/C environment outside the isolator. From there, the sample is picked up for further processing (see Figure 2).
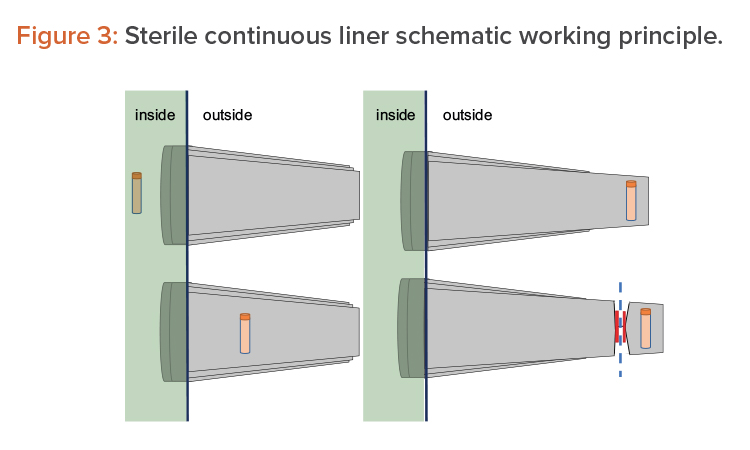
Another option consists of using pass-through boxes (also called “mouse holes”) for easy product exit. The final option is to use continuous liners and a form of rapid transfer ports (RTPs), which can be connected and disconnected due to their sterile connection mechanism. On the isolator side (i.e., in the Grade A environment), it appears as a traditional RTP. The beta container is replaced by a sterile polyethylene long tube, folded into itself several times. The object to extract is placed on the bottom of the tube, as shown in Figure 3 (1, 2). Then the tube is elongated from outside, as shown in Figure 3 (3), and welded or crimped to seal it with respect to the internal environment. The bottom part is then cut away together with the sample/product to be taken out, as shown in Figure 3 (4).
Waste management
Waste management differs a lot between the two approaches and deserves special attention. Waste management has strict rules that must be respected, and it tends to occupy a lot of the available space, drastically reducing the capability of having free working space available. Moreover, it is a potential source of particles, which impacts what may otherwise be a safe and clean environment, increasing the intrinsic risk of failures.
The types of waste can be different: it can be liquid waste used with culture media and plasticware. Typically, you produce a lot of plasticware during a C> process; some is clean and some is potentially contaminated, e.g., used flasks, tubes, pipettes.
In cleanrooms, there are different bins available to stow the materials while the batch is in process. In isolators, specific solutions have been implemented to keep waste under control. A dedicated collection chamber, is connected to the main chamber by a passage, and it behaves as an exit chamber, with all the interlocking and decontamination capabilities.
Once filled, it can be emptied and further used, providing hydrogen peroxide features. One of the best waste solutions is the continuous liner already discussed for samples. For liquid waste, there typically are vacuum pumps and a sealed bin in the technical area, which can be sent to directly disposal, or specific bottles to collect the whole batch liquid waste to be disposed at the end of the process.
Innovation
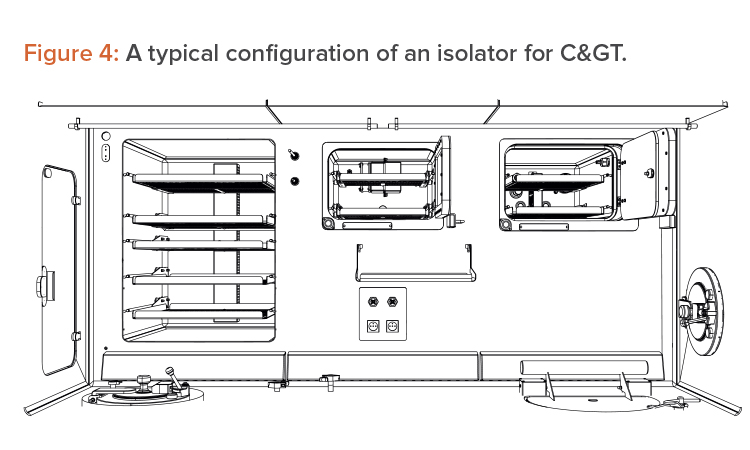
The isolator approach forces innovation and suggests improvements in space management and in device integration. Isolators force an efficient use of the available Grade A space, so movable trays have been introduced, to allow alternative use of the workbench, as well as an intensive use of the back and the lateral walls (which is not allowed in a BSC). These are all fundamental steps in gaining space, as shown in Figure 4.
Different devices used for cell processing can be integrated, even commercial devices such as a commercial centrifuge. This type of device integration keeps operators out of the Grade A working space and eliminates useless parts in the device itself (e.g., the engine and the electronics of the centrifuge). This is a fundamental step for space management, for heat management, and to avoid having moving parts inside the Grade A working space wherever possible.
In Figure 5 (a, b, c, and d), the integrated device (a centrifuge) is the sole bucket, with rotor facing the Grade A environment. The original lid has been removed and adapted. The centrifuge body, engine, and electronics are under the workbench, and the devise is coupled with a shock absorber rubber band (white) and an inflatable gasket to seal the lid while in operation.

Automation
An additional benefit in the use of isolators is the possibilities coming from the integration of different kinds of automation. Isolators are native-software-controlled machines that offer the addition of different levels of interactions: at the lower level, there are sensors and monitoring systems; at the same level, there are other isolators and related equipment; and at a higher level, there are operator interfaces (e.g., standard operating procedures [SOP] monitors and company enterprise resource planners [ERP]).
As an example, we worked with the architecture of the controlling system of a plant with three isolators, internal incubators, refrigerators, freezers, and centrifuges; a set of 144 external mobile incubators (and corresponding automatic transport system); and 20 VPHP generators.
Dedicated communication between resources has been foreseen to allow local data exchange and coordination of activities, so the VPHP generators talk with isolators, the isolators talk together and with incubators, the automatic transport system (FMS – Flex Management Software) talks with incuba-tors and isolators. By accessing the common communication bus, the different components of the system talk with high-level software interfaces and databases, like SOP monitors and company ERP.
Discussion
Isolation has proven to be an efficient and reliable technology in many fields of the pharmaceutical business, and its use is now widely accepted. Its application in C> is still limited, compared to traditional pharmaceutical environments, but the situation is changing and more C> applications are added daily to the number of those working according to isolation technology.14
We have reviewed the basics of integrating a C> process born in cleanroom under isolation technology, and we have demonstrated the possibility of successfully carrying out the process, together with the necessary steps, tools, and adaptations the isolators need to have for managing complex processes in a similar way to C>.13
An increased use of isolation technology in C> is introducing additional challenges, both in terms of new manufacturing technologies to be integrated, and in terms of different ancillary operations to be managed and performed. At present, the main manual activities have been “moved” from an open approach to the closed approach under isolators, and we discussed the difficulties encountered but also the positive outcomes of the analysis performed on the single tasks and the solutions adopted for a smooth and flawless transition. We believe that the next step will be the integration of C> in dedicated production processes under isolation technology, where the positive benefits of the same (improved assurance of quality, reduced footprint, increased sustainability) can be integrated with the benefits of automation.
This discussion shows that complex processes can be operated in an isolator under Class A conditions with resultant improvements in quality assurance and the potential for reducing operating costs. Not discussed yet is that isolators have the potential to be more portable than cleanrooms, which are very challenging to make portable. This presents a completely new opportunity, as today C> manufacturing is mainly centralized in a relative few number of plants. This is a great advantage for isolators allowing manufacture of C> products to be carried out closer to clinic sites and making speed of manufacture faster.
Conclusion
Although historically many C> projects have been initiated in cleanrooms, isolation technology is now mature enough to allow its use in C> applications. In fact, several processes can now be successfully transferred under it. These two approaches are different, and many aspects need to be evaluated. For example, there are no fixed rules, except those coming from regulatory bodies, and no lasting and constant situations because they can change from time to time or from product to product.
The proper application of an in-depth process, pharmaceutical engineering knowledge, and robust regulation can lead to the solution and reinforce the potential superiority of the closed environment approach. The implementation is not simple, but many issues, as described here, have already been addressed and there is a validated solution available.



