Implementing Lifecycle Validation Practices at Contract Manufacturing Organizations

This paper discusses the nuances of lifecycle validation implementation at contract manufacturing organizations (CMOs). Much has already been written on the general implementation of best practices for lifecycle validation, including the elements of quality by design (QbD).
1 Introduction
This paper discusses the nuances of lifecycle validation implementation at contract manufacturing organizations (CMOs).1 Much has already been written on the general implementation of best practices for lifecycle validation, including the elements of quality by design (QbD). 1,2,3 CMOs have unique considerations for lifecycle validation implementation, however. These include differentiating responsibility for various stages of lifecycle validation for both the CMO and the customer, implementing quality systems that allow flexibility for various customer approaches, process knowledge‐ transfer mechanisms between the CMO and the customer, and generating a quality agreement that captures the elements of lifecycle validation.
1.1 Overview
This paper discusses how CMOs can establish unique working partnerships to implement a process validation (PV) lifecycle approach for active pharmaceutical ingredient (API) and drug product production.
Life cycle stages discussed in this paper are:
Process design: The process is defined, based on knowledge gained through development and scale‐up. The sending site should plan knowledge transfer, the use of communication tools, and how cross‐ company teams will use and interpret process fitting. 4, 5
Process qualification: The process design is evaluated to determine if it is capable of reproducible commercial manufacturing. This typically includes developing an approach to evaluate equipment selection and qualification, and agreeing on the approach to process validation and production of the initial PV lots. 6, 7
Ongoing process verification: Confirms that the process remains in a state of control during routine
production and post‐commercialization. 7, 8
1.2 Agreements, Confidentiality, and Intellectual Property
A confidentiality agreement should be in place prior to starting any project. It should be reviewed and accepted by all stakeholders. The agreement should cover nondisclosure of partnerships that have not been made public. It should also detail how intellectual property (IP) will be used, what use of IP is prohibited, and whether IP will be applied to competitive products or used to generate competitive products. The agreement should also indicate if IP rights will extend beyond the related process or product.
IP may be related to:
- API supplied by the MAH
- IP developed under contract with the CMO
- IP retained by the CMO on processing or techniques
The contract should also include quality assurance (QA) and commercial agreements. The cost of implementing the contract should be stated clearly.
A transfer agreement may be required to allow the MAH to share product information, ingredients, or other IP‐protected materials as part of the transfer. The agreement may include a non‐use provision. Before sharing any documents, team members should consult with legal.
2 Process Design
2.1 Knowledge Transfers
A knowledge transfer is typically required to allow process design data from MAH development activities to be understood and adapted for the establishment of critical process parameters by the CMO. It should include detailed information on deliverables (including raw materials), development work (sometimes in a database), storage and transport, cleaning, analytical, and regulatory aspects. The details maybe loaded into a shared file, but the knowledge should be transferred in a way that is understood and confirmed by the personnel performing tasks.
One way to accomplish this is to detail the process in action‐based maps (flow charts) developed by the working and quality assurance teams. Maps should highlight product‐specific activities and give special attention to the rationale for changes. It is the MAH’s responsibility to ensure that information provided to the CMO is clear and concise. While coherent communication is always required for technical transfers, it is essential between the MAH and CMO, as the costs for failure increase significantly as the stages progress.
A process design plan should use a risk‐based method to define critical quality attributes derived from product and process knowledge, clinical trials, and other data. A production process should be based on design (e.g., QbD) data and any historical information available. The MAH and CMO should work together to provide developmental information, including goals of different lots that were produced, parameters that were adjusted, and effects on the process or product. Risk evaluation will also improve process understanding and help identify critical process parameters. The MAH should send retained samples of development batches to the CMO for evaluation and confirmation that both analytical and production results are comparable.
Development data should be managed adequately (preferably in a matrix database) and provide the basis for the technology transfer. Data should include information on raw material, processing, and analytical variants.
Assess analytical methods against current standards, regulatory standards, and their applicability to the product. If the matrix database contains significant data points from each batch, use look‐up tables to compare key data. A gap assessment to compare critical process parameters (CPPs) and critical quality attributes (CQAs) can help determine risk‐reduction methodology. Scale‐up may require additional strategies and proof that the process will meet the verification requirements.
When sending samples, in‐process materials, and finished products between sites, shipping condition testing should be conducted to determine the environmental and physical effects of shipping on the product. Shipping condition testing is especially important if the material will be stored, shipped, or distributed across different climatic zones or if seasonal changes may affect the product.
Providing a process design summary report and raw data tables to the CMO will facilitate the technology transfer. The report should include a defined manufacturing process, completed CPP/CQA matrix, completed risk evaluation, control strategy plan, risk‐reduction plan, test‐method variability, and statistical assessments of available pilot, engineering, and control data. Process variation to date should be identified and used as a predictor for validation readiness. Ideally, before proceeding to validation batches a process should demonstrate control, with critical process values falling within predefined limits and data indicating expected distribution.
When transferring production capabilities to another site, a skill set assessment can measure the training and knowledge of CMO personnel against the product requirements. High risk gaps should be addressed with a training plan; this may include having MAH personnel train CMO personnel.
2.2 Quality Agreements
EudraLex Volume 4, Chapter 7 defines the requirements for outsourced activities in the European Union and defines responsibilities of both the MAH and CMO. 9 These include assessing the legality, suitability, and ability of the CMO to conduct the outsourced activities; establishing the quality agreement; and reviewing CMO performance. The quality agreement should ensure that all arrangements for outsourced activities are agreed upon by both parties and are in compliance with regulations and marketing authorization for the product concerned. The quality agreement should also describe clearly who is responsible for each step of the outsourced activity.
2.2.1 FDA draft guidance
In May 2013 the US Food and Drug Administration (FDA) released draft guidance on quality agreements in contract manufacturing arrangements for drugs. 10 The guidance, similar in scope and approach to EU good manufacturing practice (GMP), describes how the MAH and CMO should delineate their roles and responsibilities to assure drug quality, safety, and efficacy. The FDA and other health authorities consider CMOs to be extensions of the MAH, therefore both are considered accountable. Health authorities also state that the quality agreement “should clarify which of the CGMP [current good manufacturing practice] activities are to be carried out by each party.” 10Health authorities do not typically review the business contract (master service agreement); it is essential, however, that it aligns with the quality agreement, which the health authorities often will review.
2.2.2 Roles and responsibilities
The roles and responsibilities of the MAH and the CMO may not be the same as those required for products transferred from one CMO's site to another CMO's site. At least some of that difference may be based on risk.
To facilitate the utilization of a lifecycle (or any validation) approach, responsibilities for generating and approving both technology transfer plans (including analytical methods) and validation or qualification plans should be defined clearly in the quality agreement. Because specific activities and responsibilities will depend on the individual product, ownership, level of process understanding, etc., these details should be addressed in the appropriate plans and protocols, not in the quality agreement.
Technology transfer and process validation plans are typically approved by the MAH to assure concurrence with the proposed strategy and responsibilities, as well as any regulatory considerations. The quality agreement should require the CMO to approve equipment, facilities, and utilities, and to have a master plan or procedure in place. For equipment, facilities and utilities, while the quality agreement should require the CMO to qualify these and to have a master plan and/or procedure in place, this is often reviewed during visits or audits of the CMO rather than through formal approval of the plan.
CMOs often take a similar approach with validation or qualification of computer systems and software used in GMP‐related activities associated with the product. The quality agreement should require that the CMO has procedures in place to assure the integrity, archiving, and retrieval of electronic data to comply with applicable regulations, but these do not require MAH approval.
The quality agreement should also detail responsibility for analytical methods development and validation. Some pharmaceutical companies rely on the API provider to provide assay methods and on compendial pharmacopeial monographs 2 to provide standardized methods and specifications for generic pharmaceutical raw materials and finished products. Selecting an acceptable analytical method selection is typically the MAH’s responsibility, but the group that performs testing (typically the CMO) is responsible for understanding it and assuring that test results are both accurate and repeatable. Selecting, transferring, and validating the analytical method may take as long as the process transfer.
2.2.3 Change management
Change management is a key element of the lifecycle approach. The quality agreement should require that the CMO have an approved written procedure for control of changes affecting the product. These include:
- Manufacturing components and raw materials
- Manufacturing process
- Packaging materials
- Labeling
- Manufacturing equipment
- Facilities and utilities
- Computer hardware/software
- Product, raw material
- Shipping
- Specifications
- Auditing raw material suppliers
- Test methods
The change procedure should ensure that changes are appropriately evaluated and approved by the CMO quality unit (QU). The change control process may shift between the CMO and the MAH, depending on the nature of the manufacturing agreement. An MAH providing ingredients to the CMO should require change‐control information to be passed to the CMO when changes occur. Changes to ancillary processes that affect the product (such as the water system) may require the CMO to complete product‐related risk assessments. Information on the changes should be readily available when batch release occurs.
The change procedure should also include the process and criteria for MAH notification and approval, follow‐up, and closure. Evaluating changes and their effects on the contracted product are a key step in the process, and both parties should understand how these changes will affect the manufacturing process in the long term. The quality agreement should require that significant changes affecting product identity, strength, safety, potency, purity, stability, regulatory status, or validation/qualification are communicated in writing.
The MAH and CMO should agree on how a significant change will be evaluated. Changes to equipment, a material, or an operation affected by the control strategy could then be considered a notifiable change or deviation. The MAH should also specify if they expect to be notified about a change that would affect product quality on a positive as well as a negative basis. A control strategy and risk assessment can help the MAH and CMO agree on what level of risk would require notification. Both parties should also agree on a timeline for change request submissions once the need for change becomes apparent, with sufficient time for the customer to comment and approve or reject changes prior to implementation. Provision for emergency changes can also be referenced.
A master batch (or device) record is a living document established during product development and confirmed through validation activities designed to ensure that the process for product manufacturing delineates appropriate controls. Control of the master batch record (e.g. change control, who approves, and when) should be detailed in the quality agreement.
In addition to change management, the quality agreement should require the CMO to have a system to control specifications including: raw materials, product labeling, packaging materials, and other materials that would likely affect product quality. These responsibilities should be agreed upon and understood, particularly where each party has different responsibilities for raw material supply. Some MAHs may toll the API to the CMO, for example, while the CMO sources other components directly. Control of raw materials is critical to quality; loss of control in a process is often caused by a raw material change. The key is to establish and maintain a firm foundation of raw material control for the validated process.
A common practice is to have MAH subject matter experts (SMEs) present during development, validation, and the initial commercial production run. In some high‐risk scenarios, the SME may be present into the commercial stage of the lifecycle. This can be a source of confusion and doubt if roles and responsibilities are not clearly defined in the quality agreement and commercial contract.
Some commercial contracts include sections that allow the MAH to have a "man in the plant”:
Not with‐standing the foregoing MAH has the right of access to CMO Facility consistent with the QAA. MAH shall have the right for at least two (2) persons to act along as “man in the plant” concept in CMO Facility. Duties and Responsibilities of this man in the plant are to oversee the Product is Manufactured in accordance with the Specifications.
Such terms on should be defined clearly, as the CMO may be unwilling to have the MAH oversee their personnel; there may also be issues related to scheduling.
On the positive side, the MAH SME should provide insight and direction when questions or problems arise. Definitions of and limitations on subcontractors, outsourcing of sourcing materials, or services should be addressed in the quality agreement. This is not intended to reduce a site’s abilities, but to ensure timely communication and planning, and ensure product quality. The CMO should have procedures in place for qualifying subcontractors that provide GMP‐related materials and services, including provisions for an appropriate written contract and auditing. The quality agreement should require that subcontractors involved in the production, holding, and release of products are approved by the MAH before implementation, and also make clear that the CMO is as responsible for the acts and omissions of permitted subcontractors as if those acts or omissions had been carried out by the CMO itself.
2.3 Quality by Design (QbD)
Agreement on process design approach (e.g., a risk‐assessment method, parameter‐classification approach, process‐characterization plan, etc.) between the MAH and the CMO can be challenging, since the MAH likely has a defined approach consistent with other projects or regulatory submissions, while the CMO may be balancing varying requirements between multiple MAHs. This can lead to differing approaches to QbD within similar design spaces.
It is important to note that MAHs should not impose their quality systems on a CMO just because they are different. The agreement in a QbD approach, or lack thereof, may be more or less significant depending on whether or not the CMO is assisting with process development, which may include formal process‐characterization studies. (CMOs may have a bench scale process [where applicable] that is use initially for technology transfer, and later for process fitting and characterization.) From a CMO perspective, it is important to have quality systems in place that either govern (if internal) or facilitate (if external) QbD best practices. ICH guidance is clear that the majority of QbD is a regulatory expectation (in countries where ICH applies). Because they are subject to regulatory review and inspection, the CMO is obliged to comply with regulatory expectations and not merely assume that QbD compliance is the responsibility of the MAH alone.
A CMO’s best option is to implement systems that facilitate QbD while ensuring that such systems are open enough to allow for a variety of MAH needs. Defined procedures, for example, should be in place for parameter risk assessment, scaled‐down model qualification, and process characterization (where applicable). Such procedures, however, should allow for flexibility in specific methodology to define design space parameters, if applicable. Rather than specifying that parameter risk assessment should be performed via failure mode effects analysis (FMEA), for example, a CMO procedure on risk assessment may call out the various risk assessment tools identified in ICH Q9, Annex 1 11 and then suggest circumstances in which certain risk‐assessment tools are more relevant than others.
The MAH and the CMO should work together to adopt the appropriate QbD tools that meet the project needs. Typically, the MAH is responsible for quality target product profile (QTPP) and CQA identification; these should be identified before the technology transfer to the CMO. If, however, the CMO is codeveloping a product, for example, then procedures to define the QTPP and CQAs may need to be in place.
The essence of process design—definition of a process control strategy—depends heavily on process knowledge that resides both with the CMO and the MAH. While scientific knowledge about the product and perhaps the process, are predominantly “owned” by the MAH, equipment capability, level of automation, alarming, batch record writing, etc. are primarily “owned” by the CMO, so this information should be available in a format that facilitates the QbD activities required during process design. For QbD to be a successful joint venture between MAH and CMO, it is beneficial to have this type of information compiled and available in source documents (e.g., historical production data reports, equipment capability summary report, etc.).
3 Process Qualification
3.1 Equipment and Facility Qualification
The process owner (MAH) has a responsibility to verify that their CMO has an adequate qualification program in place and needs to identify any gaps from both a capability and a qualification standpoint. The CMO is responsible for establishing and maintaining their facility and equipment in a qualified state (i.e., showing that it is fit for intended use), and for successfully managing any facility and equipment regulatory obligations and inspections. Establishing that the facility and related process equipment are qualified to meet the needs of the new process may best be performed as part of a process‐specific evaluation and risk assessment. This will also identify any areas of high risk to the product.
One method for this evaluation is to engage the CMO in a process‐assessment exercise such as an FMEA. When performed to an appropriate level of detail, this exercise will confirm that capabilities and process controls are in place, or identify any area(s) needed for improvement. Key aspects for review are existing process controls including alarm strategy, deviation handling and process intervention, sampling capabilities, and data recording and retrieval (e.g. historian). Once the high‐risk or deficient areas are identified they can be assessed for resolution that may include process modifications, equipment changes or qualification, or additional engineering controls.
Qualification requirements of the facility and equipment assessment allow the existing qualification program and its components to be evaluated. This identifies any gaps in existing qualification for subsequent remediation. It is important to note that the CMO’s qualification strategy may not align with the MAH’s, although both traditional IQ/OQ/PQ (installation, operational, and performance qualification) and risk‐based ASTM E2500 12 approaches are acceptable. It’s also important not to impose the MAH’s systems just because they are different. What is critical is that an adequate and comprehensive validation program is in place, and that it includes acceptable qualification practices and change control.
To ensure a comprehensive assessment, it’s crucial that the MAH and CMO commit to a control strategy and risk assessment as part of the quality agreement. This is a common project challenge as performing an assessment to the required detail often takes time. But this activity confirms the CMO facility’s equipment and processes, ensures that the facility is fit for the intended use, and identifies high‐risk aspects that should be addressed. After unacceptable risks have been mitigated and facility capabilities confirmed, the CMO should be ready to initiate the new manufacturing process and support associated process validation batch execution.
The cleaning validation process includes additional considerations when producing products at a CMO, because both MAH and CMO need assurance that cross‐contamination will not occur. MAH contracts may require the CMO to list the type of products produced at the facility, as well as written notification when a new type of product is introduced. CMOs require safety data sheet and cleaning information from MAHs—including available cleaning procedures or documented cleaning studies of the compounds—before they bring materials on site. When safety and cleaning information is not available in early‐stage development, a conservative approach should prevail. This may include dedicated equipment, disposable materials, and various engineered or personal safety systems to protect personnel from exposure. MAHs typically require assurance that foreign materials (e.g., detergents, extraneous ingredients) do not contaminate the finished product.
3.2 Initial Process Validation Batches
3.2.1 Working with the customer to agree on number of initial PV batches required and additional studies
To generate product that meets predefined requirements every time, it’s in both the MAH’s and CMO's interests to ensure that validated processes are robust. A team of experts from both the MAH and CMO can use data gathered from process design to determine the level of control in the process. The team may include members from technical, project management, R&D, quality control, quality assurance, supply management, procurement, regulatory (license transfers, etc.) and other disciplines. Teams work with clearly defined scope, milestones, and communication plans to ensure timely progress updates. A responsibility‐assignment matrix (RACI diagram) identifies who is Responsible for each activity, who is Accountable that activities are completed according to requirements, who must be Consulted on activities, and who is Informed about the activities (see Figure 1).
Figure 1: RACI Diagram
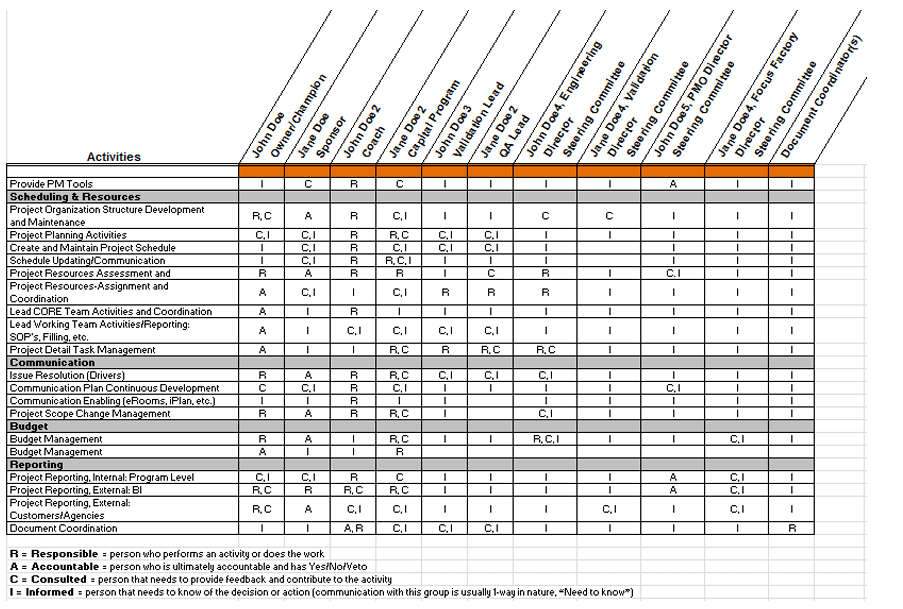
Process design data is often developed with scaled equipment and verified with commercial‐scale engineering batches. The questions to be asked are:
- What has changed?
- How do we demonstrate control of the change?
- Can data from development batches created at MAH be used to demonstrate control at the CMO?
Data can often be combined if controls were maintained across both sites and if the data is representative of the defined process. Agreement between the MAH and CMO on the number of batches required to demonstrate control and the documented rationale for that number should be based on current industry and regulatory expectations. 13, 14 Current FDA and EMA regulatory expectations emphasize that the level of monitoring and testing performed during initial process‐ validation batch studies (process‐performance qualification) should be sufficient to determine if the process is capable of reproducible commercial manufacturing.
A statistical method for determining the number of samples to satisfy initial PV batch requirements should be used as appropriate. While not required, statistical methods can also be used to determine the number of qualification runs required.
The monitoring and testing scheme required to show that a pharmaceutical batch process is operating in a validated state can be defined using a combination of risk assessment, control charting, and/or capability statistics. A science‐ and risk‐based plan—in conjunction with statistics, as appropriate— should be developed to ensure quality and to justify commercial release. ISPE has published multiple articles identifying development of a rationale for the number of batches required. 15, 16
A process risk assessment developed in process design can be used to select appropriate levels of statistical confidence and coverage to support initial PV criteria measurement. Determining process performance capability is an ongoing process that should continue as part of process qualification until control is fully demonstrated. Once process qualification milestones have been met, commercial distribution batches can be released, and testing and monitoring may be reduced to commercial production levels.
A joint MAH–CMO team may create a validation summary report using the outputs from equipment and facility qualification supporting lifecycle validation and initial PV. The report summarizes the information and data demonstrating that the commercial manufacturing process is capable of producing acceptable quality products consistently and within commercial manufacturing conditions.
The MAH and CMO should address stability and transport validation studies. Stability study requirements vary depending on the product, anticipated shelf life, and regulatory agencies in specific regions of distribution. Transport validation studies should be completed with in‐process and finished products that are susceptible to changes caused by transport; they should reflect worst‐case conditions to be effective. Realistic transportation conditions can be identified using previous history, geographic distribution zones, and transport studies.
4 Codevelopment of ongoing monitoring strategy
Developing the ongoing monitoring strategy should begin with the completion of the process validation summary report once the initial validation batches have been completed. Ongoing monitory strategy may include a review of the MAH product annual report (if commercial product was transferred), as well as applicable CMO annual report data to ensure that triggers for issues identified in the past are monitored by the CMO and confirmed by the MAH. This review should include three steps:
- Review the MAH annual product report process steps to identify reported issues with relevant root cause identification.
- Review the CMO's applicable process steps that indicate a potential process step issue and relevant root cause issue.
- Ensure the identified triggers that caused the issues are recorded to demonstrate control.
The control strategy developed in process design defined CPPs to be monitored and adjusted for batch production in commercialized product. These should be confirmed or modified when the initial PV is completed. The quality or commercial agreements may include responsibilities for identifying trends and appropriate actions during batch production and assessing batch‐to‐batch production trends. Variation and risk reduction goals for the MAH–CMO team may also be established to implement continuous improvement goals.
Commercial production should continue to demonstrate that the commercial process is operating in a state of control. This can be demonstrated by individuals, moving range, and range/sigma charts in conjunction with capability statistics, as appropriate. MAHs typically review data from the first batches when they are produced and may move to a less frequent review after both the CMO and the MAH agree to an established frequency.
The quality agreement defines the expectations for batch production based on the market authorization requirements. Under CGMP requirements, the CMO’s QU is responsible for approving or rejecting products or services provided under a contract. 17, 18 Agreements may state that the MAH has final batch disposition authority and may request notification when certain boundaries have been breached. A drug product assay that has always trended around 99–101%, for example, may result in MAH notification when a new batch has a 103% assay. Some contracts include notification bands to alert the MAH when an out‐of‐trend (but in‐specification) result has occurred. The goal is not to reject or accept a batch, but to understand the inputs and controls to ensure the patient is always protected. Some agreements for highly technical production processes include a section detailing when adjustments should be administered. The Nelson rules provide guidance on when to make adjustments.19
Process improvements require science‐based design and understanding to be realized. Sources of variation should be identified and categorized, and experiments designed to benefit both the MAH and CMO should be considered for investment by both parties. Some contracts include a section rewarding consecutive variation reduction, and provide specific realistic targets for production processes (e.g., average assay values of 100% [± 0.5%] for monthly production and batch content uniformity values to reach a process performance index [PPK] of > 1.33). Compliance to requirements is critical for every batch to ensure patient safety. Demonstrating process control across all batches provides supply chain security and makes financial sense.
Batch data is combined in the product database and can be used to identify trends such as effects of tooling wear, seasonal changes, and raw material differences. All of these may be used to identify changes quickly using principle component analysis; this type of analysis may also expand the QbD CPPs. Because CPPs are associated with key performance indicator values, they allow the team to visualize the direct effect of process changes on the product. By normalizing the values and applying product, process, and analytical knowledge, the team can continue to improve production processes so that technicians can quickly differentiate normal process variation from process shifts. Understanding the causes of process shifts—and preventing them—will ensure the state of control.
Process improvement program results may become intellectual property, depending on how they were developed and their long‐range effects. MAH commercial contracts may specify that process improvements developed by the CMO during the development of the product are the MAH's intellectual property. This means, for example, that if the CMO identifies a way to increase output, the MAH has the right to patent the information because it was developed during the production of the MAH's product.
Risk analysis should be used to determine the extent of changes and their effects on the overall process qualification. Commercialized lifecycle products that are transferred from a MAH to a CMO should follow a plan similar to the one outlined above.
5 Conclusion
Both the MAH and CMO should work together when developing, implementing, and maintaining product lifecycle validation activities. Guidelines provide a structural overview of requirements, but the individual product and patient needs is the cornerstone of any lifecycle product development and implementation. Requirements should be established during development, documented in the appropriate specifications, and adhered to using procedures defined by the quality agreement. Successful implementation requires both parties to share information according to a well‐defined quality agreement.
Limitations and Liability
This paper discusses the nuances of lifecycle validation implementation at contract manufacturing organizations (CMOs). Much has already been written on the general implementation of best practices for lifecycle validation, including the elements of Quality by Design (QbD). CMOs have unique considerations for lifecycle validation implementation, however. These include differentiating responsibility for various stages of lifecycle validation for both the CMO and the customer, implementing quality systems that allow flexibility for various customer approaches, process knowledge‐ transfer mechanisms between the CMO and the customer, and generating a quality agreement that captures the elements of lifecycle validation.
Please direct all feedback to pvpapers@ispe.org.
Acknowledgments
By Authors: Robert Beall (ProPharma Group), Penny Butterell (Pfizer), Kurtis Epp (CSL Behring), Lois Hintz (Corden Pharma), Russell Miller (Lilly), Rusty Morrison (Commissioning Agents), Mike Westerman (IPS) The authors would like to thank Gretchen Allison (Pfizer), Joanne Barrick (Lilly), and Jennifer Walsh (BMS) for all of their hard work and support.


