Industry Perspectives on Practical Application of Platform Analytical Procedures

Pharmaceutical and biotechnology companies employ platform analytical procedures in the development stages of their synthetic and biological drug products and are beginning to leverage them for commercial products. This shift is supported by the acceptance of platform procedures in the recently adopted ICH Q2(R2) and ICH Q14. Six case studies are shared in this article to highlight how platform procedures are developed, applied to products in development, and assessed for extent of validation needed to determine if appropriate for use.
The recent revision to ICH Q2(R2) Validation of Analytical Procedures1 and new guideline ICH Q14 Analytical Procedure Development2 have provided a pathway for pharmaceutical and biotechnology companies to develop and use platform analytical procedures for commercial products.
ICH Q2(R2) defines a platform analytical procedure as “an analytical procedure that is suitable to test quality attributes of different products without significant change to its operational conditions, system suitability, and reporting structure. This type of analytical procedure can be used to analyze molecules that are sufficiently alike with respect to the attributes that the platform analytical procedure is intended to measure.”1
Background
Platform analytical procedures have been commonly used across industry to streamline clinical development by making analytical development and validation activities more efficient. The inclusion of the platform analytical procedure concept in ICH Q2(R2) formalizes the idea in guidance for the first time. ICH Q2(R2) states, “when an established platform analytical procedure is used for a new purpose, validation testing can be abbreviated, if scientifically justified.”1
ICH Q14 further describes the rationale for application of platform analytical procedures and reduction of subsequent development activities: “In certain cases, an analytical procedure can be applied to multiple products with little or no modification of measurement conditions. For a new application of such platform analytical procedures, the subsequent development can be abbreviated, and certain validation tests can be omitted based on a science- and risk-based justification.”2
The Food and Drug Omnibus Reform Act (FDORA)—a US Food and Drug administration (FDA) “rider” that is part of the Consolidated Appropriations Act, 2023—required the US FDA to establish a program for the designation of platform technologies.3 It states that the sponsor of a drug application should demonstrate that the platform technology has the potential to be incorporated in, or utilized by, more than one drug without an adverse effect on quality, manufacturing, or safety. Although FDORA applies to manufacturing technologies, some concepts may also relate to platform analytical procedures, which this article aims to explore.
To date, industry has used the platform analytical procedure approach primarily for biological products and nearly exclusively in clinical development.4, 5, 6 However, recent research applied a platform approach to the analysis of residual solvents in small molecules7 and has offered suggestions for submitting information on analytical platform technologies in regulatory filings.5 Monoclonal antibodies (mAbs) are good candidates for platform analytical procedures. This is because most attributes can be analyzed by an established set of conditions where the mAbs are of similar size and structure.
In a recent guideline from the World Health Organization (WHO), platform technologies were defined for methods and processes as they apply to mAbs.8 Similarly, messenger RNA (mRNA) vaccines are good candidates to adopt platform analytical procedure strategies, as they may have common structural elements with only the codon-optimized sequence encoding the target antigen being unique to each new mRNA construct/variant. Moreover, many platform analytical procedures with established conditions, system suitability, and reporting structure have been used to analyze mRNA attributes for a commercial product.9
The article showcases examples from six different companies to demonstrate the efficiencies and opportunities afforded by platform analytical procedures across techniques and modalities, leveraging prior knowledge and historical data. Although a pathway has been recognized through the adoption of ICH Q2(R2) and Q14, challenges lie ahead in preparing and gaining approval for global submissions.
By sharing these examples, the aim is to provide awareness of and create dialogue around global acceptance of platform analytical procedures. The objective of this article is to describe how the implementation of platform analytical procedures benefits industry and regulators alike.
Pfizer: Platform Analytical Procedures for mRNA Vaccines
The following case study shows the use of platform analytical procedures to support mRNA vaccine technology. All analytical procedures supporting re-lease and stability testing for an mRNA COVID-19 vaccine were fully validated in conformance with existing ICH guidelines. Platforms have been established through extensive method development, validation, transfer, and testing of mRNA vaccine products—and their effectiveness was demonstrated through an application to change an mRNA strain for the already approved product.
The approach outlined in this example can be used for any mRNA vaccine product or product change by following the appropriate assessment, described next.
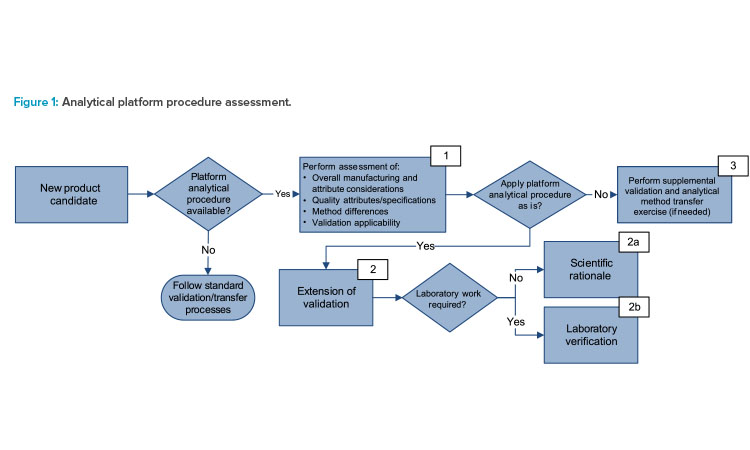
The critical elements of a procedure and the rationale to support any reduced validation requirements are determined using a science- and risk-based approach. Figure 1 shows a decision tree used to apply the strategy with consistency and considers the following elements in box 1:
- Overall considerations of the manufacturing processes, matrices, and attributes
- Quality attributes and alignment of acceptance criteria (specification) to the analytical procedure’s intended use
- Changes to analytical methodology, reference materials, and critical reagents
- Applicability and coverage of the validation, e.g., validated range and transfer activities
The assessment described in box 1 of Figure 1 concluded that there is no change for the changed mRNA product other than the sequence in the platform manufacturing process, the target concentration, and the specification. The analytical procedures have already been established at the commercial testing sites. Next, one example of each outcome—scientific rationale (Figure 1, box 2a), laboratory verification (Figure 1, box 2b), and supplemental validation (Figure 1, box 3) and the rationale are given.
By sharing these examples, the aim is to provide awareness of and create dialogue around global acceptance of platform analytical procedures.
Extension of Validation: Scientific Rationale
For box 2a in Figure 1, RNA concentration by ultraviolet (UV) spectroscopy was selected as the example. The sequence change from mRNA-to-mRNA products does not alter the basic structural components—the purine and pyrimidine bases that absorb UV—of DNA or RNA. This allows for determination of RNA concentration by the Beer–Lambert law when measuring absorbance at 260 nanometers (nm).
The UV spectroscopy platform procedure was previously validated per ICH Q2 and transferred to the testing site. No changes to the platform analytical procedure are required to support testing of the new product. The procedure could be run without impacting system suitability criteria, operating conditions, and parameters.
Extension of Validation: Laboratory Verification
For box 2b in Figure 1, mRNA integrity (purity) using capillary gel electrophoresis (CGE) was selected as the example. Considering no significant change in size/valency, the sequence length of the new mRNA product/strain may affect migration time and the electrophoretic profile of fragments and intact species without impacting the ability of the method to accurately measure RNA integrity.
Verification was performed to confirm that the potentially impacted characteristics of the platform analytical procedure remain in a validated state. For this example, precision and specificity were challenged under a protocol with the same acceptance criteria. The CGE platform procedure was previously validated per ICH Q2 and transferred to the testing site.
No changes to the platform analytical procedures are required to support testing of the new product. The same assay control, sample preparations, and instrument conditions are used. The analytical procedure is applied without significant change to its operational conditions, system suitability, and reporting structure.
Supplemental Validation
For box 3 in Figure 1, identification of mRNA using digital droplet polymerase chain reaction (ddPCR) was selected as the example. The procedure requires mRNA product/strain-specific primers and probes (for specificity). Challenge validation characteristics (e.g., specificity) were considered under a protocol with acceptance criteria against both the formulation buffer and other mRNA constructs. The ddPCR platform procedure was previously validated per ICH Q2 and transferred to the intended testing site.
Platform Documentation for Regulatory Submissions
Although there are multiple ways to document this work within a company, the documentation within a regulatory submission should be considered early in the process. The full validation for each analytical procedure is a combination of the platform procedure validation, scientific rationale, and product-specific data.
For any extension of validation or supplemental validation leveraging historical knowledge and/or previous validations, a validation report or summary in the appropriate common technical document sections should be provided. This includes the scientific rationale justifying the strategy for each analytical procedure. In addition, product-specific results from laboratory verifications and supplemental validations should be shared within the submission.
The mRNA technology presents an application of the platform analytical procedure approach for an already approved product upon a variant change. The same principles can be applied to other products generated with a similar manufacturing process (for example, a new mRNA vaccine candidate). The subsequent case studies aim to expand the approach to other modalities.
Genentech: Capillary Electrophoresis Sodium Dodecyl Sulfate (CE-SDS) Methods
Workflow
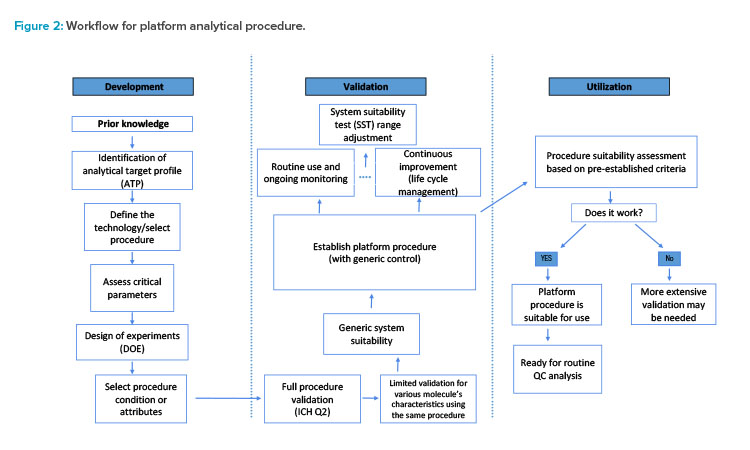
The platform analytical procedure workflow consists of three phases: development, validation, and utilization (see Figure 2). Measurement requirements for a given quality attribute or a set of quality attributes can be defined in an analytical target profile (ATP). This predefined approach to analytical development can ensure that the analytical procedure is robust across its life cycle.
To achieve the optimal procedural conditions, a design of experiments (DOE) for screening of critical method parameters is executed. During the second phase, a validation strategy is devised per ICH Q2. To ensure suitability of the platform analytical procedure for new molecules, an assessment along with a generic control is completed.
The platform analytical procedure control is used for system suitability testing, which ensures the proper performance of the analytical procedure during assessment. The platform procedure is monitored to maintain performance and prevent potential drifting through the system suitability criterion. Using a common control for system suitability across multiple labs, instruments, and analysts enables collecting more relevant data and improves the precision assessment of the platform analytical procedure. Furthermore, the control enables evaluation of any changes introduced by suppliers, such as column resins or equipment configurations.
Application
CE-SDS analytical procedures are commonly used to evaluate the size heterogeneity, purity, and manufacturing consistency of biologics, under non-reduced and/or reduced conditions. The non-reduced CE-SDS method separates, detects, and quantitates distribution of product-related size variants (e.g., hinge fragments, main peak, or partially reduced cysteine-linked subunits). Reduced CE-SDS separates and quantitates light chain (LC), non-glycosylated heavy chain (HC), HC components, and other size variants such as non-specific polypeptide cleavages (e.g., LC and HC clips).
CE-SDS is a good candidate for a platform analytical procedure due to wide applicability and well-established robustness for usage in testing of mAbs. During the developmental phase, a combination of single- and multi-factorial studies are completed to identify critical method parameters, minimize the method variation, and increase applicability of usage as a platform across multiple mAb formats.
This approach was applied to the determination of critical dye-labeling conditions for a fluorescent-based platform CE-SDS method.10 Critical parameters such as SDS-protein complexation, dye labeling, gel-lot variation, and auto-sampler stability are confirmed during validation.
The conditions for platform analytical procedures are established by leveraging prior learnings and knowledge from previous product-specific and platform-validation studies. During the utilization phase, the feasibility of the platform analytical procedure for new mAbs is evaluated through limited assessment studies, such as precision, specificity, stability indication, and comparison of UV and fluorescence electropherograms to ensure proper dye labeling.
Inclusion of a preestablished platform assay control during the assessment studies ensures proper performance of the analytical procedure. This approach creates a rigorous process to ensure that new molecules that meet the predefined criterion during the utilization phase can be tested by the platform CE-SDS method.
Eli Lilly and Company: Capillary Electrophoresis to Investigate Fragmentation of MABS
As previously mentioned, reduced and non-reduced CE-SDS are standard techniques employed in the purity assessment for large molecules. The following example offers another approach to establish a platform analytical procedure for CE-SDS analysis of size variants. The principle of the CE-SDS separation for both conditions—reducing (the addition of 2-mercaptoethanol [BME] to break disulfide linkages) and non-reducing—is based on the use of an electric field to separate proteins through a polymeric gel matrix based on their hydrodynamic radius.
The objective of this article is to describe how the implementation of platform analytical procedures benefits industry and regulators alike.
SDS is a negatively charged ionic detergent used in sample preparation to promote denaturation and achieve uniform protein coating. This is to obtain a uniform mass-to-charge ratio. Under such conditions, molecular hydrodynamic volume is the determining factor, resulting in a size-based separation. “Ignoring” the diversity in the complementarity-determining regions that make mAbs unique allows for common method conditions to be applied to mAbs of similar size and structure.
US Pharmacopoeia (USP) <1053> provides guidance and procedures used for characterization of biotechnology-derived articles by capillary electrophoresis.11 That chapter is harmonized with the corresponding chapters in Japanese Pharmacopoeia (General Information 4) and European Pharmacopeia <2.2.47>.12, 13
Process
Four platform analytical procedures are established (reducing and non-reducing conditions with pressure or electrokinetic injection) based on prior knowledge and experience with allowable ranges around the main parameters. When a new molecule enters the portfolio, experiments (e.g., exploratory DOEs) are conducted. This is to confirm that the two main factors found to be molecule-
dependent—heating time and temperature—fall within the established allowable ranges for the platform analytical procedures. The same is true for slight alterations of other method conditions that are covered by the underlying dataset used, e.g., minor adjustments in BME concentration.
Molecule-specific instructions for sample preparation that differ from the platform analytical procedures text are documented. The analysts are pointed to this documentation when executing the platform analytical procedure. If more significant modifications were needed to achieve the intended separation (e.g., changes to gel buffer detergent, reductant, wavelength, polarity, separation voltage, or instrument model), non-platform method conditions would have to be developed. These changes would result in a molecule-specific procedure.
Based on scientific evaluation (e.g., matrix components, analyte concentration, and selectivity need), a decision is made to use the pressure or electrokinetic injection platform analytical procedures. They are used throughout development for final drug substance/drug product, as well as in-process and characterization testing. The procedures are stability indicating, consistent with global regulatory guidance and industry expectations for mAbs.
During the clinical stages of development, method qualification for the platform analytical procedure is abbreviated. Linearity across the purity range, extrapolated linearity, accuracy, and matrix specificity are not executed. The only experiments performed are those to determine precision/intermediate precision, prepared sample stability, and stability indication using a stressed sample.
Molecule-specific procedure codes are then introduced at the time of marketing authorization. The validation exercise employs a mixed model of using historical data, generated across the platform, in addition to molecule-specific data. Robustness parameters are determined based on platform knowledge: the experiments conducted for each molecule.
Amgen: Leveraging a Platform-Size Exclusion Chromatography (SEC) Analytical Procedure for MAB Analysis
Purity methods, such as SEC, are ideal candidates for platform execution. SEC can be consistently applied across different mAbs due to its versatility and ability to handle diverse protein samples. SEC methods developed for one mAb can often be transferred and applied to others without any changes to the method.
This makes SEC a convenient and time-saving approach when analyzing different mAbs within a project or across multiple projects. SEC is compatible with a wide range of sample matrices, including different formulations, buffers, and excipients. It can handle mAbs derived from various sources (e.g., mammalian and microbial) or subclasses (immunoglobulin G (IgG)1 and IgG2). This versatility allows for the use of SEC as a consistent platform method across a mAb portfolio.
By selecting an appropriate column, laboratories can achieve optimal resolution and separation for different mAbs to generate a platform procedure, ensuring accurate and reliable analysis. SEC is extensively used in quality control laboratories for mAb analysis. Its ability to provide quantitative information about mAb size distribution, aggregate content, and purity makes it a reliable method for routine analysis of different mAb samples for both re-lease and stability purposes. By using SEC as a platform method, laboratories can streamline their analytical processes and maintain consistent performance.
Method Development
Typically, the development of analytical procedure commences after preclinical development for new active substances. Method development should align with the expectations of an analytical platform, which encompasses the ability to monitor relevant quality attributes, attribute sensitivity, and reporting intervals. The initiation of qualification and robustness experiments should occur after finalized method conditions and initial system suitability criteria are established.
The completion of initial robustness is a prerequisite before the method can be used for release and stability testing of clinical materials. As illustrated in Figure 3, if the method is applicable for platform use, the initial qualification and robustness lay the foundation for implementation. After the platform method is adopted in the laboratory, the method can be applied to a new molecule with minimal method qualification and robustness to demonstrate that the method is fit for purpose.
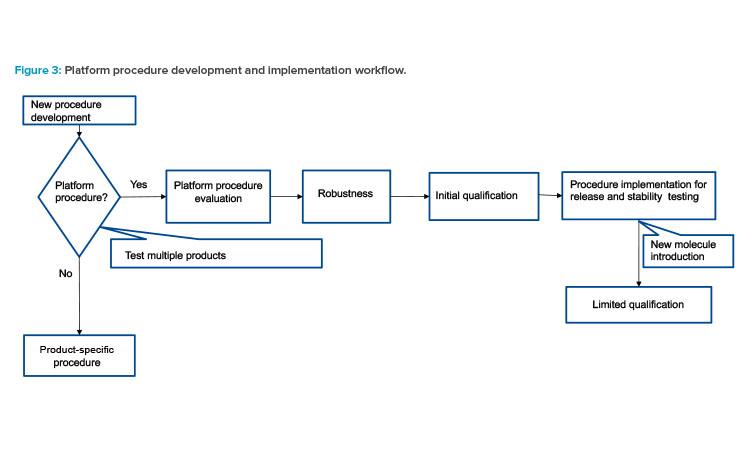
Having a subset of method qualification parameters tested prior to new product introduction is generally adequate in development. If prod-uct-specific information, such as example chromatograms and integration events, is needed to facilitate attribute testing of the new molecule, it can be achieved by maintaining specific supplemental documents. For regulatory submissions, platform and any product-specific qualification data can be provided to justify use of the platform procedure.
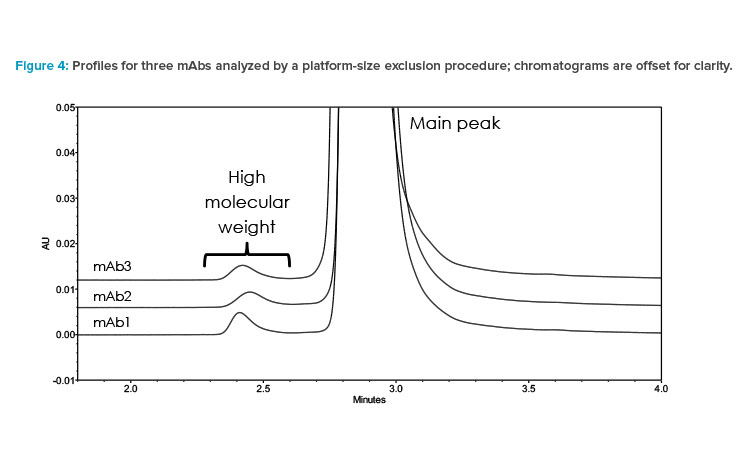
In summary, SEC’s compatibility, transferability, and suitability for diverse mAb samples make it an excellent candidate as a platform methodology. Its consistency and reliability contribute to efficient and robust analysis across different projects, as shown in Figure 4. It facilitates comparability studies and quality control and ensures the product quality of mAb-based therapies.
Sanofi: Peptide Mapping as a Global Platform Analytical Procedure
Peptide mapping has been developed and implemented as a platform procedure for identity testing of mAb and mAb-like molecules to support lot release at Sanofi. Despite being a comparative and platform procedure, peptide mapping is not a “generic” method. A specific map, or a fingerprint, needs to be developed for each protein under the same/similar operational conditions, system suitability, and reporting requirements. General guidance and procedures for this method are provided in USP <1055>: Peptide Mapping14 and harmonized with corresponding chapters in the Japanese Pharmacopeia <G3-3-14> and European Pharmacopoeia <2.2.55>.12, 13
Platform Procedure
The platform procedure consists of disulfide reduction under denaturing conditions, cysteine alkylation, buffer exchange, enzymatic digestion, chromatographic separation of peptide fragments with UV detection, and data analysis. Identity is confirmed based on a comparison of the chromatographic profile and the relative peak area ratios of selected marker peptides of a test sample against a reference standard.
To ensure the platform procedure is robust and fit for its intended purpose, an enhanced approach—as delineated in ICH Q14—was used during development. Sample preparation (from reduction to digestion), separation and detection of peptide fragments, and data analysis are three major parts of the procedure. The potential variations that may occur in the first two parts are known to introduce artifacts or impact the method performance (e.g., chemical modifications, miscleavage, and poor resolution of peptides), and thus may lead to invalid results.
The risks associated with performance were assessed based on prior knowledge using a systematic approach (e.g., failure mode and effects analysis [FMEA] or Ishikawa diagram), and risk probability numbers were generated for the various method parameters. The method operable design region (MODR) was then defined based on the results from three representative mAbs (two IgG subtypes) using DOE and one-factor-at-a-time experiments. These MODRs are expected to be applicable to many other mAbs. Thus, the platform approach affords reduced method development for new mAbs. For molecules that behave differently and do not fit the platform, a product-specific method will be developed.
Specificity, as a key requirement for identity, was assessed based on the overall chromatographic profile and through a set of purposely selected marker peptides. The finalization of marker peptides was based on ultra performance liquid chromatography with ultraviolet detection and mass spectrometry (UPLC-UV-MS), e.g., quadrupole time-of-flight MS detector, data from the target molecule, and their chromatographic behaviors. The development of the peptide map generally requires the identification of most, if not all, peptide fragments by MS.
To prepare the platform procedure for release testing of new mAbs, an abbreviated qualification (e.g., only the specificity performance characteristic with a limited number of samples) is performed for early-stage programs. A validation is completed with additional parameters considered (e.g., inter-mediate precision, column lot-to-lot variations) to support late-stage programs. The robustness data from development may be included into the validation report as ICH Q14 states. The platform procedure has been implemented for multiple mAbs and mAb-like molecules across different UPLC instrument platforms and data analyses applications.
Merck & Co: Platform Gas Chromatography Procedure for Residual Solvent Quantification in Small-Molecule Pharmaceuticals
Organic solvents are commonly used in the production of small-molecule pharmaceuticals, primarily for drug substance and occasionally for formulated drug product. Ensuring that the levels of residual solvents are within acceptable limits, as defined by ICH Q3C (R8),15 is crucial for both clinical and commercial release. Therefore, it is essential to analyze and control the organic solvents used in the manufacturing process.
Gas chromatography with flame ionization detection (GC-FID) is the preferred technique for quantifying residual solvent levels. This is due to its compatibility with solvents of different boiling points, availability of various stationary phases for different selectivity, separation capability of capillary columns, and the universality and sensitivity of FID.
Analyzing Residual Solvents
When analyzing residual solvents with GC-FID, two main factors to consider are sample preparation/introduction (direct injection or static headspace sampling) and chromatographic procedure. The chromatographic procedure typically employs single capillary columns and an oven temperature program for baseline resolution of the analytes.
USP <467>: Residual Solvents,16 which is harmonized with European Pharmacopoeia Chapter 2.4.24, Identification and Control of Residual Solvents,13 describes three headspace GC-FID procedures that employ capillary columns and static headspace injection. These procedures use hydrogen, nitrogen, or helium as the carrier gas for the determination of solvents. They have a chromatographic runtime of approximately 60 minutes and use capillary columns with different phases and coating thickness as well as temperature programs.
During development, the drug substance process undergoes frequent changes and optimization to enhance cost, yield, and process robustness. This necessitates the rapid analysis of samples produced under diverse conditions. It involves conducting solvent tests for isolated intermediates at different stages of synthesis and for the final drug substance. Having a platform GC-FID procedure simplifies analysis for residual solvents under these varying conditions, minimizing method development, accelerating key results, and reducing validation activities.
Developing a Platform GC-FID Procedure
A platform GC-FID procedure based on direct injection has been developed and validated using five different diluents to achieve this goal.7 The procedure uses hydrogen as the carrier gas and can resolve over 30 commonly used solvents within a short runtime (eight minutes), which also makes it amenable to high-throughput analysis.
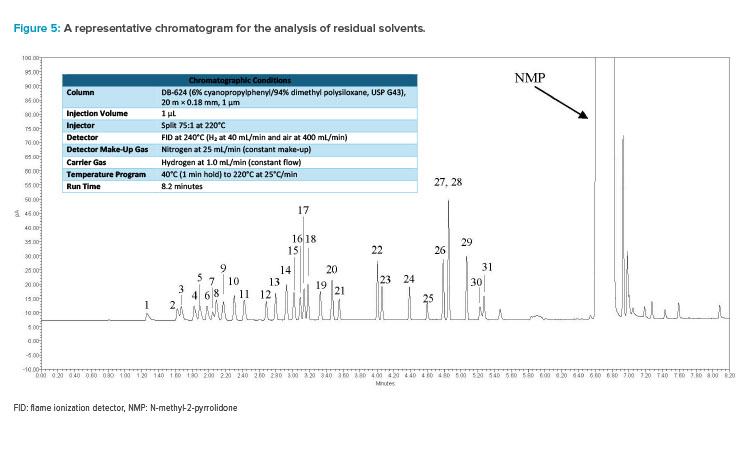
The procedure was established by conducting extensive development and validated using standards for sample independent attributes, such as line-arity, sensitivity (limit of detection/limit of quantitation [LOD/LOQ]), and precision. Various clinical-stage compounds, approved drug substances, and commercially available materials were used as model compounds to assess matrix effects, and the method’s accuracy was demonstrated through spike and recovery experiments for solvents likely to be present in the respective compound (see Figure 5 for a representative chromatogram and Table 1 for procedure details).
The platform GC-FID procedure is commonly used during development of small-molecule programs at Merck & Co. When considering a new program in early clinical development, the suitability of the platform procedure is evaluated based on its ability to separate the desired solvent(s) from process impurities and sample matrix interferences (specificity). Additionally, the stability of the sample solution is assessed to ensure an appropriate analysis window after preparation. If the peak resolution and solution stability results are acceptable, the method can be implemented without the need for further validation.
For commercial programs, supplemental validation of the platform procedure has been performed for application to drug substance. This includes accuracy assessment through spike/recovery, intermediate precision, and robustness evaluation using specific solvents of interest. Additional pro-gram-specific requirements are determined through a case-by-case assessment, and prior knowledge of the platform procedure is leveraged to support validation studies as needed.
Analyte Number | Analyte | Analyte Number | Analyte | Analyte Number | Analyte |
|---|---|---|---|---|---|
1 | Methanol | 11 | n-Propanol | 21 | 1,4-Dioxane |
2 | Ethanol | 12 | Ethyl Acetate | 22 | Toluene |
3 | Diethyl Ether | 13 | Tetrahydrofuran (THF) | 23 | i-Butyl Acetate |
4 | Acetone | 14 | Cyclohexane | 24 | n-Butyl Acetate |
5 | i-Propanol | 15 | i-Butanol | 25 | Dimethylformamide |
6 | Acetonitrile | 16 | Isopropyl acetate (IPAc) | 26 | Ethyl Benzene |
7 | Dichloromethane | 17 | 2-Methyltetraydrofuran | 27,28 | p-Xylene/m-Xylene |
8 | t-Butanol | 18 | n-Heptane | 29 | o-Xylene |
9 | Methyl tertiary-butyl ether | 19 | n-Butanol | 30 | Dimethyl sulfoxide |
10 | n-Hexane | 20 | Methylcyclohexane | 31 | Dimethylacetamide |
With accelerating clinical development, streamlining analytical activities without negatively impacting the assessment of product quality becomes essential.
In many cases, no further validation may be needed to apply this method to regulatory starting materials, intermediates, and in-process testing. Using a platform procedure for residual solvents has significantly accelerated process development and reduced validation requirements for introducing new small-molecule compounds to clinic as well as for commercialization.
Discussion and Example Comparison
Six companies shared examples that highlight how platform procedures are developed, applied to products across modalities in development, and assessed for extent of validation needed to demonstrate fitness. The two CE-SDS examples illustrate that multiple approaches are suitable to establish platform analytical procedures for the assessment of the same quality attributes.
Although one example was applied to commercial registration of mRNA analytical procedures, the remaining examples describe use of platform procedures for the clinical development of mAbs and small molecules. However, consistent with ICH Q2(R2), the principles described in these examples can be applied to commercial products beyond the modalities discussed previously.
Each example leveraged a science- and risk-based approach and followed similar approaches to those outlined in the presented flow charts. Extensive prior knowledge is employed in the development of the platform analytical procedure. This consists of knowledge of the modality as well as requirements of the measurement, analytical technique, and procedure parameters. Measurement requirements for the quality attribute(s) can be described in an ATP, which guides technology selection. An evaluation can be performed if the platform analytical procedure meets the performance requirements of the ATP.
ICH Q14 states, “prior product knowledge plays an important role in identifying the appropriate analytical technique. Knowledge of best practices and current state-of-the-art technologies as well as current regulatory expectations contributes to the selection of the most suitable technology for a given purpose. Existing platform analytical procedures (e.g., protein content determination by UV spectroscopy for a protein drug) can be leveraged to evaluate the attributes of a specific product without conducting additional procedure development.”2 A risk analysis is completed to evaluate if the platform can be applied to a product with or without any modifications, then the extent of validation experiments required can be determined.
Benefits of Platform Analytical Procedures
There are many benefits to the application of platform analytical procedures, particularly in the commercial environment. With accelerating clinical development, streamlining analytical activities without negatively impacting the assessment of product quality becomes essential. Platform analytical procedures can enable rapid support of new products during development and subsequent commercialization. This also leads to an increased probability for enhanced robustness. They offer operational, compliance, and training advantages for analysts that do not need to learn unique procedure parameters for every test of every product.
Beyond a reduction in validation activities, transfer activities may be streamlined where testing is consolidated in the same commercial testing laboratory. Additionally, platform analytical procedures provide opportunities for automation that can increase throughput. These advantages may lead to greater reliability of the supply chain, providing value to companies, regulators, and patients. Using the same procedure parameters and validation results additionally leads to more rapid completion of registration documents and inspection readiness. This increases efficiency for health authorities during review and inspection where the procedure has previously been registered and implemented.
Once the product and procedure are registered, there are additional benefits throughout the product life cycle. Continuous monitoring of analytical procedures in the commercial environment is a resource-intensive, yet necessary, activity. Continually monitoring a platform across multiple products is more efficient and increases detectability of performance issues requiring mitigation. Management of this knowledge is critical due to the larger amount of data for one procedure and should lead to rapid and effective troubleshooting with any issues that may arise during routine use.
Although there are many benefits to the use of platform analytical procedures, there are challenges moving forward with this approach in commercial registrations. ICH Q2(R2) allows for validation testing to be abbreviated if scientifically justified. However, the extent of studies required to satisfy health authorities from non-ICH members may delay global acceptance of platform procedures and realization of their benefits. Applicants would also need to address documentation concerns, as the validation results would be provided in the dossier of another product. How that information is linked or copied for a new product would be of concern to the applicant.
The analytical procedure control strategy for the platform, including system suitability, and specific control and/or reference materials used to ensure acceptable performance should be considered. Also, the risk analysis for why a platform analytical procedure was applied to the new product and the extent of validation required would need to be communicated. Perhaps the most challenging aspect would be addressing the change management strategy.
The need to change parameters in a platform procedure for one product would require assessment for all other products that use the platform. These concerns may be reason for some applicants to reconsider or delay registering platform procedures. However, the six examples provide an excellent overview of the science- and risk-based approach that the authors believe are representative of the principles described in ICH Q2(R2) and Q14.
Conclusion
The recent adoption of ICH Q2(R2) and ICH Q14 supports the potential for registering platform analytical procedures. This publication describes six examples that outline varying yet scientifically sound approaches for developing, applying, and implementing platform procedures. Primarily, these examples have been successfully used in clinical development, but the principles can be applied to commercial registration.
Applying platform analytical procedures from one product to another has many benefits that facilitate efficient pathways to registration. Beyond registration, a more streamlined path for postapproval changes is important for the life cycle management of the product. With increasing pressures of cost, speed to market, and product quality, the use of analytical procedure platforms is one aspect of a science- and risk-based strategy for product commercialization that will ultimately benefit patients today.
Acknowledgments
The authors would like to acknowledge the ISPE Product Quality Lifecycle Implementation (PQLI)® Analytical Methods technical team for its contributions to the subject matter discussed in this paper, and David Michels, Anjana Patel, and Matt Sampson for their support with manuscript preparation and review of the publication.
Conflicts of Interest
All authors are employees of their respective companies. All authors contributed to the writing of the report, approved the final version of the manuscript, and agreed to submit the manuscript for publication.








