Toward a Single Global Control Strategy: Industry Study

During the past decade, industry has experienced a proliferation of regulatory divergence regarding the interpretation and implementation of International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) guidelines (and control strategies) across geographic regions. This article shares data that highlight instances where well-established ICH regulatory members diverge from ICH quality guidance in their evaluation of the same scientific data in chemistry, manufacturing, and controls (CMC) regulatory documents submitted by industry. The data illustrate instances when regulatory divergence led to modifications to control strategies that in turn led to multiple regional and local control strategy variants globally. A common understanding of the degree of divergence and impact is an important step toward improved global harmonization of control strategies and will ultimately benefit regulators, industry, and patients globally.
Scientific and risk-based approaches in pharmaceutical development were first explicitly described in ICH Q8 and further elaborated in ICH Q9, Q10, and Q11.1, 2, 3,4 Conceptually, quality by design improved confidence in the quality of pharmaceutical products, enhanced scientific understanding, and demonstrated the robustness of the manufacturing process. It also encouraged continuous process improvement for manufacturing. A primary incentive for industry to follow quality by design guidance is to establish a common foundation for continual improvement through global regulatory concordance for new applications.
Lately, rather than truly harmonized regulatory expectations, localized interpretations of ICH guidelines have resulted in different regulatory requirements and/or control strategies, which poses significant challenges to marketing a single product in global markets. As a result, the increased complexity of manufacturing supply chains and the regulatory burden associated with maintaining compliance with these diverse regulatory expectations have created difficulties: There are additional burdens and challenges in carrying out continuous improvement initiatives and innovation in product development is hindered. And these supply no substantive improvement in product quality, safety, or efficacy. Divergence has become a disincentive to improvements and has even caused temporary drug shortages in some markets.
Guidance and Divergences
ICH technical guidelines are used by the pharmaceutical industry to develop manufacturing control strategies. A growing number of regulatory authorities apply these guidelines to assess marketing applications to ensure pharmaceutical product quality (safety and efficacy). Applicants are consistently finding divergence in the interpretation of ICH guidelines by regulators from different countries. This suggests that determining an acceptable control strategy can be subjective.5 The metrics presented in this article—collected from core control strategy contents in marketing applications and corresponding review outcomes by health authorities—provide specific instances of divergence. This article presents metrics and examples of divergence from 112 marketing applications, covering both new synthetic and biological entities, from 11 companies from a benchmarking study conducted by the International Consortium for Innovation and Quality in Pharmaceutical Development (IQ)–Control Strategy Global Harmonization (CSGH) Working Group. The study’s objective was to evaluate the industry’s experience of divergence, build awareness of commonalities, and elucidate the implications for stakeholders (including regulators, industry, and patients). A global harmonization of control strategies should begin with a common understanding of the degree of divergence and its impact.
| Drug Substance | Drug Product |
|---|---|
| 3.2.S.2.2 Description of Manufacturing Process and Process Controls | 3.2.P.3.2 Batch Formula |
| 3.2.S.2.3 Control of Materials | 3.2.P.3.3 Description of Manufacturing Process and Process Controls |
| 3.2.S.2.4 Controls of Critical Steps and Intermediates | 3.2.P.3.4 Controls of Critical Steps and Intermediates |
| 3.2.S.4.1 Specification | 3.2.P.5.1 Specification(s) |
| 3.2.S.7 Stability | 3.2.P.8 Stability |
Data Set Criteria
Manufacturing control strategy in a submission is depicted by core common technical documents (CTDs) in Module 3 per ICH Q11. Therefore, the benchmarking survey was based on defining a set of “core documents” in a submission as a manufacturing control strategy. The core documents defining a manufacturing control strategy are described in Table 1: these include control strategy elements of material attributes, process design, in-process controls, and drug substance and drug product specifications. Study participants agreed that the control strategy described in the core documents was based on enhanced scientific understanding and in alignment with the science and risk-based approach described in ICH Q8–Q11.
Participants also agreed that, ideally, a single, complete set of core documents submitted globally is used for the survey. Companies that expect potential rejection of any core documents generally create country-specific core documents based on either previous application experience or prevailing knowledge of a country’s requirements (either explicit, published control strategy expectations, or interpretational differences). Even when core documents are submitted, a health authority may impose a revision to control strategy during application review. As a result, country-specific variation from a control strategy can be attributed to three sources:
- A company’s interpretation of country-specific regulation deems that the control strategy would not be accepted.
- A company’s experience with previous submissions that resulted in creating country-specific variant control strategy.
- Request by a country-specific regulatory authority to alter the control strategy to gain acceptance.
A global harmonization of control strategies should begin with a common understanding of the degree of divergence and its impact.
Benchmarking Data Set
After establishing a common foundation for core documents, the IQ CSGH Working Group focused on five drug substance and five drug product CTDs in Module 3, all critical in defining a control strategy, as listed in Table 1.
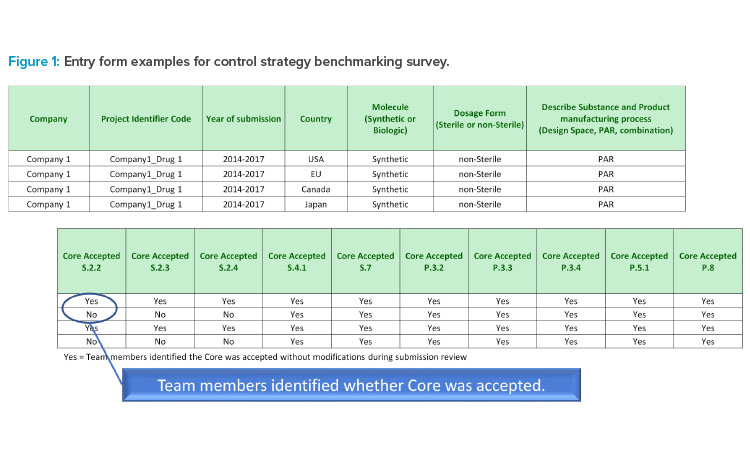
For every country where a product was submitted, study participants entered:
- Blinded company name and drug identifier
- Year of submission
- Country, market, or region (can refer to as an entity for regulatory acceptance; e.g., the EU represents several countries)
- Molecule type (biologic or synthetic)
- Manufacturing process parameter terminology used (proven acceptable range, design space, or a combination)
- Acceptance status for each of the core documents. For example:
- Enter “yes” if a core document was accepted without change.
- Enter “no” if a core document was altered prior to submission because of known regional requirements or if, during review, a change to control strategy was required to gain acceptance.
- If “no” is entered, then a description of deviation from the core document was provided and counted as the control strategy not accepted by health authority.
| Submissions | Core Document Acceptance Ratea | Average Acceptance | ||||||||||
| S.2.2 | S.2.3 | S.2.4 | S.4.1 | S.7 | P.3.2 | P.3.3 | P.3.4 | P.5.1 | P.8 | |||
| US | 30 | 63% | 67% | 63% | 50% | 57% | 87% | 47% | 53% | 23% | 57% | 57% |
| Japan | 17 | 35% | 41% | 53% | 47% | 76% | 76% | 47% | 71% | 18% | 65% | 53% |
| EU | 35 | 34% | 34% | 31% | 29% | 71% | 71% | 49% | 54% | 17% | 57% | 45% |
| Canada | 30 | 67% | 63% | 80% | 50% | 80% | 80% | 43% | 63% | 40% | 70% | 64% |
| Overall acceptance | 112 | 51% | 52% | 56% | 43% | 71% | 79% | 46% | 59% | 25% | 62% | 54% |
| Probability of acceptance by all four countriesb |
5.0% | 5.9% | 8.3% | 3.4% | 24.6% | 37.6% | 4.7% | 12.8% | 0.3% | 14.8% | 8.7% | |
a. Acceptance rate/acceptance is calculated in percentage by dividing total number of “yes” answers for core document(s) with the total number of submissions from one or multiple countries/regions.
b. Probability of acceptance by all four countries is calculated as a product of multiplication of individual core document acceptance rates of all four countries.
The entry form is illustrated in Figure 1.
The data set included information from 112 marketing application submissions from 11 companies to established ICH countries/regions: US, EU, Canada, and Japan. The data set represented 64 synthetics applications and 48 biologics applications, with 104 submitted after 2014 and 63 after 2018.
Results and Discussion
The results focus on control strategy divergence caused by different expectations of ICH guidelines between the industry and health authorities. The method used to generate metrics does not delineate details of divergence from health authorities because these details do not alter the trend and study conclusion. Using the agreed-upon criteria for core document acceptance, the average core document acceptance rate across the US, EU, Canada, and Japan is 54% for synthetic and biologic products, as shown in Table 2. Country average acceptance rates range from 45% to 64%, with the EU having the lowest and Canada and the US the highest, at approximately 60%. When translating overall acceptance rate (54%) to the probability of core documents being accepted by all four countries, a probability of 8.7% (Table 2) is found, which indicates a > 91% likelihood that at least one of the four countries will not accept a consistent control strategy/core documents.
| Submissions | Core Document Acceptance Ratea | Average Acceptance | ||||||||||
| S.2.2 | S.2.3 | S.2.4 | S.4.1 | S.7 | P.3.2 | P.3.3 | P.3.4 | P.5.1 | P.8 | |||
| US | 17 | 88% | 88% | 88% | 65% | 82% | 88% | 71% | 71% | 29% | 76% | 75% |
| Japan | 12 | 50% | 42% | 50% | 58% | 92% | 67% | 50% | 75% | 17% | 83% | 58% |
| EU | 19 | 42% | 16% | 26% | 26% | 84% | 63% | 37% | 58% | 21% | 63% | 44% |
| Canada | 16 | 81% | 56% | 94% | 69% | 94% | 75% | 31% | 50% | 50% | 75% | 68% |
| Overall acceptance | 64 | 66% | 50% | 64% | 53% | 88% | 73% | 47% | 63% | 30% | 73% | 61% |
| Probability of acceptance by all four countriesb |
15.0% | 3.3% | 10.8% | 6.8% | 59.6% | 27.9% | 4.1% | 15.4% | 0.5% | 29.8% | 13.0% | |
a. Acceptance rate/acceptance is calculated in percentage by dividing total number of “yes” answers for core document(s) with the total number of submissions from one or multiple countries/regions.
b. Probability of acceptance by all four countries is calculated as a product of multiplication of core document acceptance rates of all four countries.
To determine areas of significant divergence, the overall probability of 8.7% (Table 2) was compared with the individual core document acceptance probability for the four countries; where the probability of individual core document acceptance was lower than 8.7%, the control strategy is deemed an area of significant divergence. Using this criterion, significant areas of divergence were identified in 60% of core documents, including S.2.2, S.2.3, S.2.4, S.4.1, P.3.3, and P.5.1, as highlighted in red in Table 2, with each at a > 91% likelihood of not being accepted by at least one of the four countries. The data demonstrate that local jurisdictional considerations hamper the desired outcome of a globally harmonized control strategy. Because the EU has the lowest average acceptance rate, it also has the most impact to the calculated probability of individual core document acceptance.
Although the number of Japan submissions used in this study is lower than the other three countries/regions, it is important to include the study results reflecting Japan’s reliance on Module 1 and a separate application form, which create a single country variant of control strategies. Removing the Japan data and recalculating a revised threshold for the remaining three countries did not change the areas of divergence, nor the finding that the EU was the most divergent. Hence, including Japan in calculating the probability of acceptance for an individual core document does not change the trend of the identified significant areas of divergence, and this trend is consistent when considering the metrics separately for synthetic and biologic products.
Although the data set is slightly weighted toward synthetic products, adequate data are collected for both molecule types, which ensures the metric analyses reflect the trends objectively. The acceptance rate and probability in the US, EU, Canada, and Japan separated by molecule type, synthetic product, or biologic product are shown in Tables 3 and 4, respectively. Overall, the observed amount of divergence in control strategy is more for bio-logic products than for synthetic products.
The number of synthetic product submissions included in the data set, 64, allows for a review of country-specific acceptance rates and areas of significant divergence as presented in Table 3. Core document average acceptance rate ranged from 44% to 75% indicating significant divergence, with the EU having the lowest acceptance rate. Both the US and Canada had an average acceptance rate of ~ 70%, which tracks the combined trend. The overall acceptance rate is 61% for synthetic products, which is 7% higher than the combined trend of 54% (Table 2). The high severity of divergence of control strategy is reflected by a low probability of 13.0% for the core documents being accepted by all four countries (Table 3), translating to an 87% likelihood of at least one country not accepting the core document. Using the overall probability of 13.0% (Table 3) as a threshold to compare with individual core document acceptance rate for the four countries, the significant areas of divergence included S.2.3, S.2.4, S.4.1, P.3.3, and P.5.1 as highlighted in red in Table 3. Additional sections that were close to the threshold included S.2.2 and P.3.4.
| Submissions | Core Document Acceptance Ratea | Average Acceptance | ||||||||||
| S.2.2 | S.2.3 | S.2.4 | S.4.1 | S.7 | P.3.2 | P.3.3 | P.3.4 | P.5.1 | P.8 | |||
| US | 13 | 31% | 38% | 31% | 31% | 23% | 85% | 15% | 31% | 15% | 31% | 33% |
| Japan | 5 | 0% | 40% | 60% | 20% | 40% | 100% | 40% | 60% | 20% | 20% | 40% |
| EU | 16 | 25% | 56% | 38% | 31% | 56% | 81% | 63% | 50% | 13% | 50% | 46% |
| Canada | 14 | 50% | 71% | 64% | 29% | 64% | 86% | 57% | 79% | 29% | 64% | 59% |
| Overall acceptance | 48 | 31% | 54% | 46% | 29% | 48% | 85% | 46% | 54% | 19% | 46% | 46% |
| Probability of acceptance by all four countriesb |
0.0% | 6.0% | 4.5% | 0.6% | 3.3% | 59.2% | 2.2% | 7.3% | 0.1% | 2.0% | 3.6% | |
a. Acceptance rate/acceptance is calculated in percentage by dividing total number of yes answers for core document(s) with the total number of submissions from one or multiple countries/regions.
b. Probability of acceptance by all four countries is calculated as a product of multiplication of core document acceptance rates of all four countries.

Although the biologic product submissions (48) included in the data set are less than the number of synthetic product submissions, it is sufficient to identify specific areas of significant divergence on control strategy for biological products, as presented in Table 4. For biological products, the individual country core document acceptance rate range is 33% to 59%. The range is lower than for synthetic products, but it indicates significant divergence among the four countries. The overall acceptance rate for biologic products is 15% lower than synthetic products (61%; Table 3). The US had the most divergence observed and Canada had the least, with average core document acceptance rates of 33% and 59%, respectively. Control strategy divergence is more than that of synthetic products and is reflected by a threefold decrease to a very low probability of 3.6% for core documents being accepted by all four countries (Table 4); this means there is a > 96% likelihood that at least one of the four countries would not accept core documents. Using the threshold of the overall probability of 3.6% (Table 4) in comparing with individual core document acceptance probability for the four countries, the significant areas of divergence with lower individual acceptance probability included S.2.2, S.4.1, S.7, P.3.3, P.5.1, and P.8 (highlighted in red in Table 4). To the extreme, Japan did not accept S.2.2 core documents in any submission, translating to a 100% certainty that the S.2.2 document would require at least one additional regional version. Similarly, low acceptance rates by multiple countries reflected a close to 100% probability that S.4.1 and P.5.1 would not be accepted by at least one country.
Upon review and discussion of the data, all IQ Working Group members agreed on the common areas of divergence across both modalities for drug substance and drug product, which are illustrated in Figure 2.
The key areas of significant divergence on control strategy based on metrics align well with the areas that have the most common areas of divergence. For both synthetic and biologic products, key areas of control strategy divergence are related to specification(s), description of manufacturing processes and process controls, and control of materials. For each key area of control strategy divergence, the associated top significant divergence is summarized in Table 5 and its impact is discussed later.
| Key Area of Divergence | Significant Divergence on Control Strategy |
|---|---|
| Specification | Justification of impurity acceptance criteria. Justification of required test types, including tests considered yet omitted, in specification. |
| Description of manufacturing process and process controls | Utilization of ICH terminology and the level of details required in describing process parameter ranges. Justification of criticality of process parameters and/or in-process controls. |
| Control of materials | Identification and justification of drug substance starting materials. |
The data shows wide variation in acceptance rates and overall low acceptance rate of documents that industry believes meet ICH control strategy requirements based on their acceptability in at least one ICH region. This is concerning in light of the well-established nature of these regulators within ICH. When considering new ICH regulators and observers, it is reasonable to assume the overall acceptance rate may drop significantly. Industry has an important harmonization strategy to develop and use a single set of control strategy documents, but regional and local preferences drive a plethora of additional commitments.
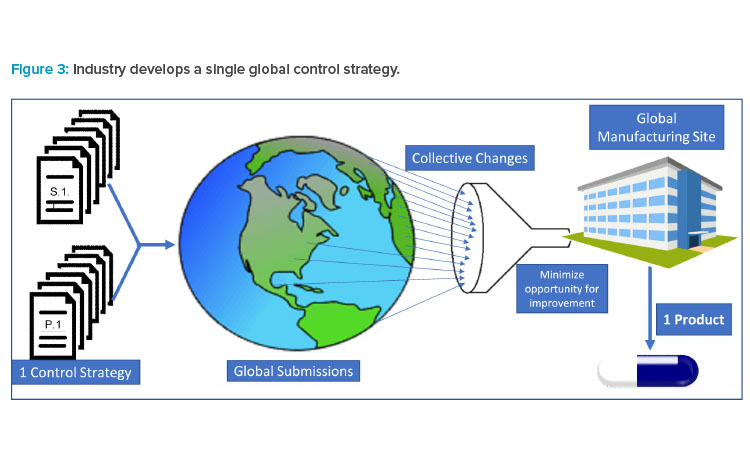
Pharmaceutical products are typically globally manufactured and released for all marketed countries, not a single or group of countries. As a result (and as illustrated in Figure 3), industry must collate all requested modifications and requirements to create a single set of requirements for “one-product manufacturing.” Industry must then accommodate regulatory requests by amending CMC controls or segregating materials for supply in a specific market.
Thus, differing accepted test methods and specifications become barriers to innovation and continual improvement for all global products and patients.
Control Strategy Divergence and Patient Impact
The lack of harmonization of control strategies delays access of new medicines to global patients. Applicants must stagger global submission plans for new medicines to allow time to answer questions from global quality regulators. Although applicants submit a single core control strategy to supply the global markets, the same science, justification, and data sets are often interpreted differently by health authorities. This difference in interpretation of control strategy suitability results in the high degree of variability in the volume of questions from global quality reviewers on the same set of documents. Frequently, an applicant will not only experience a large variation in the number of questions, but will also encounter multiple rounds of questions from a given regulator on a particular topic. Additionally, some health authorities have limited windows for an applicant to respond to questions.
As a result, subject matter experts with specific knowledge of the process, testing, and specifications must prioritize preparation of responses over other R&D efforts and spend many hours to provide additional justification for the control strategy that was submitted and/or implement control strategy changes. The time spent on these efforts limits the ability of a company to submit global applications and inhibits further development and expansion of supply that would allow global patients to have access to potentially life-saving medicines. Among the companies that participated in the control strategy survey, many observed a drastic difference in the number of questions received on the same data between agencies (for example, 19 from one agency and 184 from another). The difference in questions is all too common and reflects notable differences assessing suitability of the control strategy.
Country-specific control strategy requirements can also potentially lead to a drug shortage if material made and released for one region is unsuitable for a different jurisdiction. Pharmaceutical products are typically manufactured for global release, not for a single or group of countries. Local constraints on control strategy, such as tightened manufacturing ranges and/or specifications based on a limited number of manufactured batches, are especially costly. Alternatively, a comprehensive science- and risk-based approach is strongly favored. Although additional control strategy requirements by any given country can be accommodated, the combined requirements of over 100 countries can add significant manufacturing and supply barriers.
The lack of a single control strategy for all countries will lead to needless supply chain complexity and can have profound impact on supply of critical medicines to patients. Although companies may manufacture to the most stringent control (parameter range or specification limit) in the case of failure to meet the tighter controls, country-specific release may be applied. However, country-specific release is inherently a complex process because it is intended to be used by exception and could delay the release of product for distribution. For products with supply constraints due to manufacturing capacity, complex manufacturing process, or unforeseen supply chain disruptions, this can lead to potential stock-outs.
Sponsor X reported a case in which it was requested that an impurity specification limit, based on available batch data, be tightened, even though the process had been demonstrated to tolerate a higher limit consistent with safety considerations. In this situation, a tighter impurity limit for the intermediate would have led to a delay in availability of the new medicine in this country because the product for the launch was made with intermediate that did not meet the tightened specification. The company’s rationale for keeping the originally proposed specification was accepted, but often sponsors are forced to accept a lower limit.
ICH Considerations
Expansion of new ICH members and observers is expected to result in continued escalation of divergence and increased obstacles to realizing globally harmonized control strategies. New ICH members have the challenges of adopting ICH guidelines while building internal capability to use them properly, which creates at least temporary divergence as the health authority transitions to the ICH-enabled future state.
At the time of marketing applications, when there is limited experience and data, constraints imposed by global regulators on licensed control strategies are inconsistent with ICH and limit innovative changes after approval. One example of such limitations is how companies describe their product manufacturing controls in the marketing authorization application. Using process control terminologies—such as proven acceptable range (PAR), normal operating range (NOR), or design space (DSp)—resulted in varying interpretations of ICH guidance and led companies to abandon strict adherence to terminology to instead focus on basic scientific principles and a robust, well-controlled process. When scientific principles are applied, some health authorities insist on applying these categories in assessing applications within their jurisdiction.6, 7
Divergent interpretation and implementation of ICH guidelines among regulators is therefore a growing problem for industry. Applicants are typically left with unnecessarily constrained control strategies, which can limit shelf life, reduce process capability, and restrict changes that could otherwise be implemented through a pharmaceutical quality system. Applicants are frequently left with no option but to accept a country-specific control strategy requirement rather than risk product nonapproval or delays to approval. Examples related to selection and justifications of drug substance starting mate-rials clearly demonstrate the negative impact of such divergence.
As an example, Sponsor A proposed two crystalline products as starting materials that were justified by ICH Q11. Regions/Markets A, B, and Q did not query the sponsor’s starting material strategy; Market E was not satisfied with the proposal and requested more of the synthetic steps to be put under GMP control, stating that the proposed regulatory starting materials do not meet ICH criteria, given short synthetic routes from each starting material to the drug substance. The sponsor acquiesced to Market E, defining submissions with starting materials consistent with Market E’s preferences, creating a divergence of the control strategy that was approved in other markets. Additional drug substance process performance qualification (PPQ) requirements and other control strategy adjustments can lead to delayed approvals and delayed availability of new medicines for patients.
Similar divergence was observed in a presubmission advisory meeting. Sponsor B proposed starting materials of a synthetic drug substance consistent with ICH Q11 and sought agencies’ advice. Agency X agreed with the proposed starting materials. Agency Y did not agree with the proposal and required additional steps from the syntheses of the starting materials to be under GMP control. Agency Y’s view was that there was no way to track changes to the starting material processes in the absence of GMP control, which is inconsistent with the intent of ICH Q11. Control strategy changes were made and additional PPQ on drug substance was conducted to satisfy Agency Y. A separate marketing application with revised starting materials was submitted for Market Y, though acceptance of the original starting materials would have provided for earlier submission and approval.
Although not directly part of the survey, recent regulatory response to submissions for COVID-19 vaccines presents an interesting and positive example. Companies initially sought Emergency Use Authorization (EUA) rather than normal submission processes, and supplies were being provided from a clinical manufacturing process. The ability to gain broad approval with a single control strategy has allowed rapid distribution of the vaccines throughout the world. Had the EUA approval process been slowed by protracted control strategy queries, it is possible that access to vaccine would be limited, even now. Increasing efforts toward convergence, a collaborative review, e.g., Orbis, will improve harmonization and lessen divergence. Historically, industry CMC organizations have not held up submissions due to numerous changes to the control strategy caused by divergent interpretations of ICH guidance, but rather as more innovative technology is used to manufacture pharmaceutical products, it will become more challenging for industry to be able to accommodate local needs without resulting in delays.
Continual Improvement and Impact on Postapproval Changes
After a product has been approved by the regulatory agencies, it is standard to make improvements in the pharmaceutical manufacturing process to increase production scale, or to implement technological advancements such as real-time testing, or to modify control strategies.
Continual monitoring efforts are in line with ICH Q8, Q9, and Q10 guidance: ICH’s Questions & Answers for ICH Q8, Q9 and Q10 states:
Continual monitoring (e.g., via Continuous Process Verification) can further demonstrate the actual level of assurance of process consistency and provide the basis for continual improvement of the product. Quality Risk Management methodologies of ICH Q9 can be applied throughout the product life cycle to maintain a state of process control.8
A holistic approach to quality improvements as described in the ICH guidance is the desired state for a robust quality improvement. The FDA and EMA further describe the principles of continuous process verification and how companies may take advantage of new advancements when applying enhanced process understanding coupled with risk management tools and a pharmaceutical quality system. Application of continual improvement in the current regulatory environment is a challenge, made more so when a product has customized controls for multiple markets.9, 10
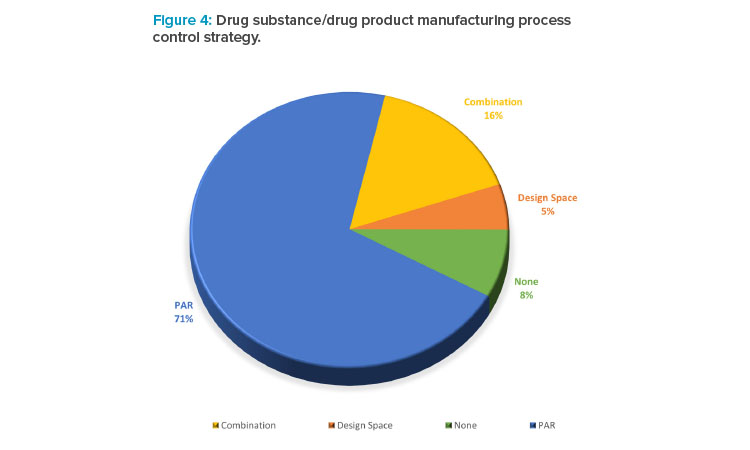
In the collected data set, companies were asked to describe their drug substance and drug product manufacturing process (Figure 4). The data illustrated that the term of design space is not used as frequently as PAR, despite most companies routinely undertaking some degree of enhanced development and studying interactions between process variables and product quality in developing the control strategy.
Companies indicated that expectations for justification and change management for control strategy features such as PAR and design space differ across regions and may not be aligned with expectations in ICH guidance.11 In addition, guidance for postapproval changes globally may categorize all changes to design space as major prior approval changes, regardless of the risk to quality. This discourages the use of design space and does not align with the concepts of quality risk management in ICH Q8–11 or with ICH Q12.12
Furthermore, some companies expressed concern that ICH Q12, which will provide guidance on how consideration of risk is used to inform which process parameters should be established conditions and how consideration of risk should inform the notification category for changes, may further confuse the use of the term “design space.”
Industry would like the focus to be on the science where production data is reviewed within the quality system for process capability and stability to drive continuous quality improvements. Unfortunately, the aggregated effects of imposed, divergent control strategy requirements by health authorities hamper continual improvement.
New medicines are typically manufactured at a single facility for global use, especially early in the product’s life cycle. Drug product batches are manufactured to a single quality standard and are designed to supply the global market. Master batch records, analytical test methods, and specifications must reflect the combined global control strategy requirements. Although control strategy requirements imposed by a single country may appear to have small impact, the combined effect of country-specific requirements is immense. For instance, Module 3 may contain nearly 40 documents and because of known or imposed control strategy requirements, the global set of documents will result in hundreds of documents to manage at the conclusion of a single life cycle review. In the collected examples, one member company cited four documents that had expanded into 24 documents approved globally due to country-specific control strategy requirements. Therefore, under current circumstances, quality improvement implementations are difficult because multiple market-specific Module 3 documents must be revised to support a given change and to meet the requirements of each market.
| Country | Submissions | Core Document Acceptance Ratea | Average Acceptance | |||||||||
| S.2.2 | S.2.3 | S.2.4 | S.4.1 | S.7 | P.3.2 | P.3.3 | P.3.4 | P.5.1 | P.8 | |||
| Combined | 112 | 51% | 52% | 56% | 43% | 71% | 79% | 46% | 59% | 25% | 62% | 54% |
| Synthetic | 64 | 66% | 50% | 64% | 53% | 88% | 73% | 47% | 63% | 30% | 73% | 61% |
| Biologic | 48 | 31% | 54% | 46% | 29% | 48% | 85% | 46% | 54% | 19% | 46% | 46% |
Although some country-specific requirements are expected, the industry strives to maximize alignment of control strategies such that the low-risk improvements can take place most efficiently and industry and regulator dialogue can be reserved for the essential elements of the control strategy. However, industry must adapt when markets have differing requirements, including postapproval requirements to support the change. These requirements may include generating additional data such as stability data prior to submission, which results in significant delays. Once global approval has been achieved, industry then implements changes to mitigate the supply chain burden and maintain supply continuity. As markets approve the change, staff members carefully track and update internal document management systems to accurately record hundreds of approvals.8 Manufacturers have found there are few degrees of freedom to operate due to the combination of specialized requirements and although the recent implementation of ICH Q12 Technical and Regulatory Considerations for Pharmaceutical Product Lifecycle Management is intended to streamline postapproval changes that require regulatory submissions, the lack of agreement on control strategy presents an obstacle that must be overcome to implement ICH Q12. As a result, optimization and improvement may continue to be limited to the few degrees of freedom to ensure manufacturers stay within the combined regulator-imposed control strategy requirements rather than adopting a holistic approach to quality improvements as described in the ICH guidance.
Future Perspectives
An overall < 9% probability for all four established ICH markets to accept a single control strategy, based on a study of 112 submissions, indicates the need for industry and regulators to work together to develop a common understanding of control strategies. This is a global problem that should be solved together; regional approaches will not address the issue. Although regulator receptivity for a global control strategy is low for both synthetic and biologic molecules, the acceptance rate of control strategy for biologics is significantly lower, as shown in Table 6.
Conventional synthetic small molecules typically require a finite amount of defined CMC content in regulatory submissions, but even here there seems to be little agreement among regulators of sufficiency criteria. Even more information is expected for large molecules such as monoclonal antibodies. Experience to date with control strategy dialogue among sponsors and agencies has been complex, but not unfamiliar to both parties.13
It is not unreasonable to anticipate ever-increasing information expectations for new modalities including oligonucleotides, live modalities, oncolytic viruses, hybrid modalities such as antibody-drug conjugates, and nanobodies.14 Given current circumstances, advanced modalities will likely present unique and unknown challenges as well as potential for greater differences within the industry and among regulators. Harmonization of new modality control strategy questions must be addressed in a more streamlined, rapid manner because the variability of health agency questions and the industry responses to these entities have added years to launching harmonization efforts.
Collaboration among industry and regulators to achieve a globally acceptable control strategy is possible and has been proven even more urgent during the current COVID-19 public health emergency. Collaboration extended beyond the ICH regions provides first steps to provide safe and effective quality life-saving medicine to patients globally.15 (For example, see the April 2020 statement from the International Coalition of Medicines Regulatory Authorities [ICMRA].) This trend toward better and more productive dialogues between regulators with mutual recognition and workload sharing is very promising, because it is essential for regulators to move toward a common scientific understanding of the core CMC information for a global product. In addition, parallel review opportunities such as Project Orbis and the ACCESS consortium can be used to drive international regulatory harmonization efforts for quality information.16, 17, 18
One possible solution to transform not only the regulatory submissions process itself, but to also streamline parallel health authority reviews is to develop a cloud-based data exchange platform for one global quality dossier.19, 20 Such a platform would improve the transparency of sponsor–regulator interactions for the CMC content across different health authorities as well as allow visibility to data packages and queries, thus encouraging commonality of technical detail while reducing redundant requests for information. Automation of CMC content and data with the use of structured content and data management would also improve submission authoring efforts and enable real-time updates and data tracking.21
Conclusion
The efforts toward global regulatory harmonization of product control strategies and CMC content are more essential now than ever before to accelerate the delivery of innovative therapeutics to millions of patients around the globe. As health authorities have pursued use of work-sharing or mutual reliance to accelerate new medicines to patients and reduce workloads, the value of a global dossier available to all global regulators has been made apparent. Despite these potential benefits to global patients, industry, and regulators, this benchmarking study revealed that country-specific requirements can emerge.
Ultimately, the IQ Working Group’s goal is to provide data and understanding for rapid improvement toward global harmonization of control strategies driving collaborative reduction in divergence and increase in harmonization, with the support of industry and regulators. This would enable accelerated drug development including novel modalities, advance innovative technologies, and ensure product supply and continual improvement through efficient lifecycle management.14 The metrics reported here are the first steps toward dialogue and solutions. The gain will be when we do a deeper dive on specific issues that are common across and engage in dialogue with health authorities. The Working Group’s recommendation is to have discussions with multiple health agencies in the near future, together, in a forum with real working solutions. The group is open to other ideas from other member companies.
Acknowledgments
The authors would like to acknowledge the International Consortium Control Strategy Global Harmonization Metrics Working Group for its collection of data and contributions to the subject matter discussed in this article. The authors thank the following individuals for their support with manuscript preparation and review: Ruth Boetzel, Nancy Benz, Sharvari Borkar, Claudia Borm, Scott Coffey, Diana Fladerer, Werner Heilmann, Kathrine Nielsen, Dennis O’Connor, Ron Ogilvie, Sophie Patton, Matt Popkin, Joerg Schiewe, Oliver Thiel, Frank Wetterich, and Kirsten Wright. The authors also recognize the support of the International Consortium for Innovation & Quality in Pharmaceutical Development (IQ).








